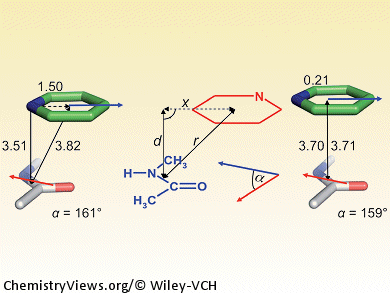
π-Stacking plays an important role in protein–ligand binding. In contrast to the aromatic–aromatic stacking of phenyl or heteroaryl rings, interactions with protein amide π systems, with their large dipole moments, is less well understood. By complementing quantum chemical calculations with data mining in experimental protein X-ray structure databases, Michael Harder, François Diederich, ETH Zurich, and Bernd Kuhn, F. Hoffmann–La Roche, both Switzerland, investigated the geometric preferences and energetics of π stacking of various heteroarenes on planar protein amide fragments. The results are relevant for structure-based drug design, as the less polar π surface of protein amides is accessible for ligand interaction in many binding sites, either as part of the protein backbone or in Gln/Asn side chains.
The authors derived some practical guidelines for rational drug design: such interactions depend heavily on the relative orientation and magnitude of the interacting dipole vectors, and less on the ideal alignment of partial charges; moreover, the π-electron density of the heteroarene correlates with binding strength. These results will help chemists establish better protein–ligand interactions through structure-based design and should also inspire further studies to explain stacking interactions between protein amide groups and (hetero)arenes.
- Efficient Stacking on Protein Amide Fragments,
M. Harder, B. Kuhn, F. Diederich,
ChemMedChem 2013, 8(03).
DOI: 10.1002/cmdc.201200512



