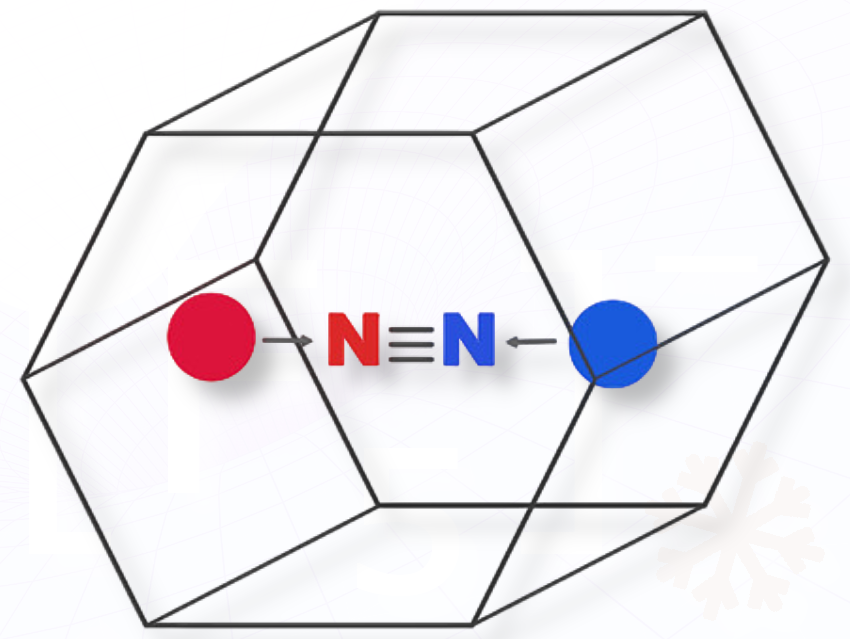
Holliness Nose, Kenya, and Fernando Ruipérez, Spain, discuss computational studies showing that molecular cage design can enable efficient nitrogen activation, which is important for developing sustainable methods to convert nitrogen into useful chemicals under mild conditions.
What did you do?
We designed and computationally studied a new family of dual Lewis acid–base molecular cages capable of binding and activating nitrogen (N₂).
These cages combine acidic and basic sites in one molecular framework so that both can interact cooperatively with N₂—one donating and the other withdrawing electron density—to weaken its very strong triple bond.
Why are you doing this?
Ammonia synthesis is still dominated by the energy-intensive Haber–Bosch process, which requires high temperature, high pressure, and metal catalysts. Finding metal-free systems that can activate N₂ under mild conditions is crucial for more sustainable “green ammonia” production and for reducing greenhouse-gas emissions.
What is new and cool about your work?
We introduce cage-like frustrated Lewis pairs (FLPs) that confine acid and base centers within one molecule while keeping them far enough apart to remain reactive. This dual-site “nano-reactor” design mimics some aspects of enzymatic pockets and transition-metal complexes but is entirely metal-free.
Our calculations reveal how geometry and electronic tuning control N₂ capture.
What is the main significance of your results?
We found that boron- and aluminum-based acids combined with strong donors like borylenes or Verkade’s bases give the best synergy for N₂ activation.
Structural flexibility and the right acid–base distance are critical: too close, and the pair neutralizes itself; too far, and cooperation is lost.
The work establishes design principles for next-generation FLP systems.
What part of your work was the most challenging?
The hardest part was balancing geometry and reactivity. Many cage structures were either too rigid or too flexible, making it difficult to keep the Lewis acid and base reactive yet cooperative.
Accurately modeling this delicate interplay required extensive electronic-structure and bonding analyses.
So, theoretical chemistry and smart molecular design are important for your work.
This study highlights how theoretical chemistry can map the design space before synthesis, saving experimental effort. It also shows that thoughtful molecular architecture, rather than heavy metals, can open new pathways for one of chemistry’s toughest challenges: breaking the triple nitrogen-nitrogen bond under ambient conditions.
Thank you very much for sharing these insights.
The paper they talked about:
- Dual Lewis Acid–Base Molecular Cages Facilitate Cooperative N₂ Activation: Insights from Theory,
Holliness Nose, Fernando Ruipérez,
ChemPhysChem. 2025.
https://doi.org/10.1002/cphc.202500624

Holliness Nose is a Lecturer and Research Scientist at the Department of Chemistry & Materials Science, Faculty of Applied Sciences & Technology, at the Technical University of Kenya (TU-K), Nairobi, Kenya.

Fernando Ruipérez is an Assistant Professor at the POLYMAT and Department of Physical Chemistry, Faculty of Pharmacy, at the University of the Basque Country (UPV/EHU), Vitoria-Gasteiz, Spain.
Also of Interest