Hydroacylation reactions can be used for the synthesis of ketones from alkenes. This approach is useful due to the good availability of substituted alkenes. Linear hydroacylation products are easily accessible via the addition of a ketyl radical to an alkene. However, this method cannot be used for the synthesis of branched addition products, because in this case, an unstable primary carbon radical would need to be formed as an intermediate.
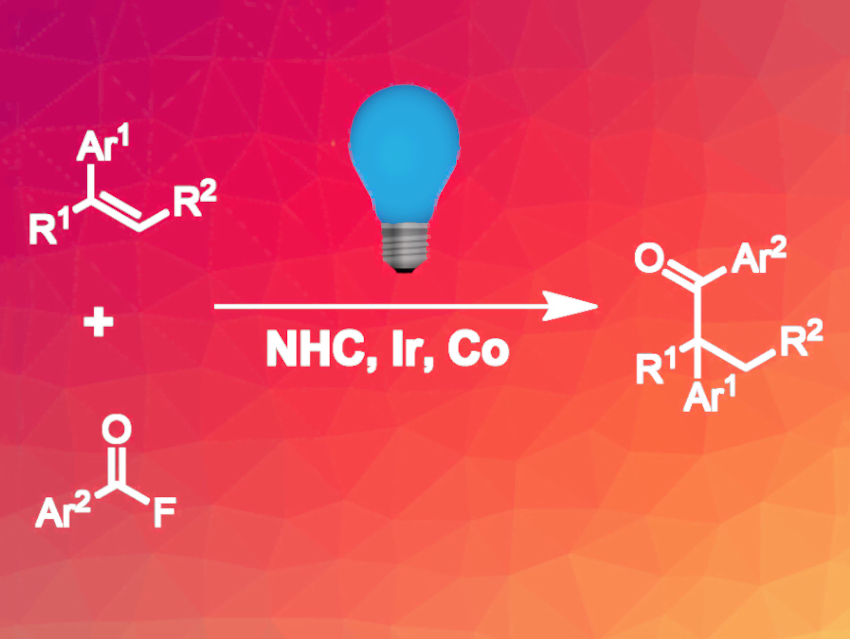
Yi Wang, Nanjing University, China, and colleagues have developed a hydroacylation protocol that gives branched ketones (pictured above). The team used a combination of a cobalt catalyst, an N-heterocyclic carbene (NHC) catalyst, and an iridium photoredox catalyst (catalysts/precursors pictured below). The procedure was optimized for the reaction of 4-tert-butyl styrene and benzoyl fluoride, which gave 91 % yield of the target product with exclusive Markovnikov selectivity. In addition to the catalysts, phenylsilane was added as a hydrogen source, and K2HPO4 was used as a base.
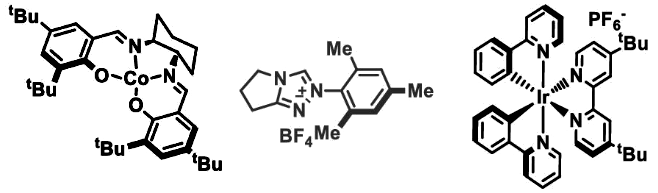
Upon irradiation with blue LED light, the Ir catalyst oxidizes the cobalt precatalyst to a Co(III) species, which reacts with phenylsilane to give a cobalt hydride species. The hydrogen atom is then transferred to the alkene, yielding a radical and regenerating the Co(II) catalyst. The NHC catalyst activates the acyl fluoride, which is then reduced by the Ir catalyst, giving a ketyl radical and regenerating the Ir(III) catalyst. The two radicals combine to form the target product.
The reaction is compatible with a wide range of different para-substituents at the styrene reaction partner. For ortho-substituted and heterocyclic styrenes, yields were lower. For the acyl fluorides, various substituents were tolerated, but compounds with heteroaryl substituents led to lower yields. The reaction was successfully applied to complex natural products and pharmaceutically active molecules.
- Branched-Selective Hydroacylation of Alkenes via Photoredox Cobalt and N-Heterocyclic Carbene Cooperative Triple Catalysis,
Xiangzhang Tao, Qing Wang, Lingyu Kong, Shengyang Ni, Yi Pan, Yi Wang,
ACS Catal. 2022.
https://doi.org/10.1021/acscatal.2c04970
![]()