Using enzymes for different reactions than those they promote in nature can be useful in organic synthesis, especially for asymmetric transformations. However, achieving the formation of C–F bonds by “unnatural” enzyme catalysis has remained challenging—in spite of the fact that organofluorine compounds are often useful, e.g., in pharmaceutical chemistry. Several nonheme Fe enzymes can promote C(sp3)–H halogenations, but so far, no conversion of these Fe halogenases to fluorinases could be achieved.
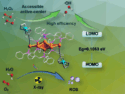
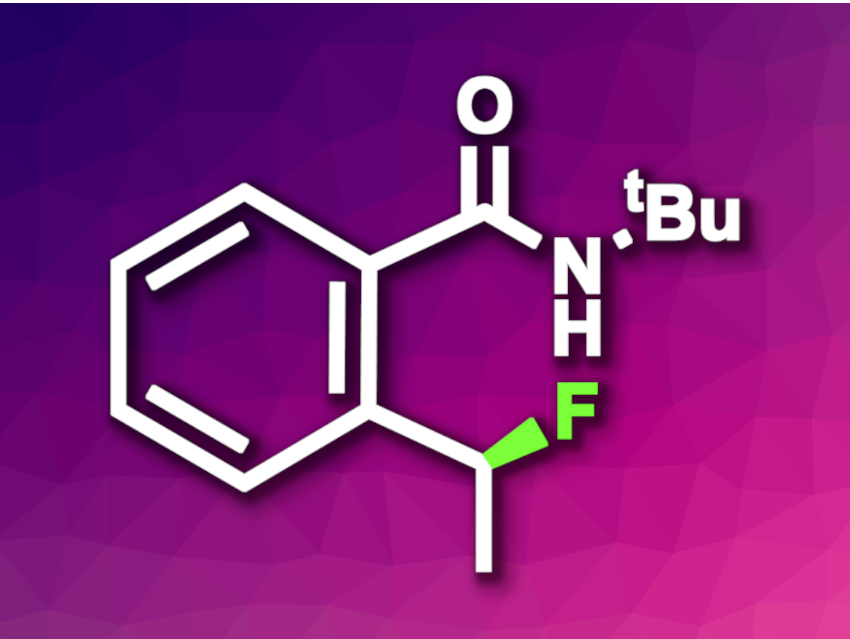
Peng Liu, University of Pittsburgh, PA, USA. Yang Yang, University of California, Santa Barbara, USA, and colleagues have used directed evolution to convert the natural nonheme enzyme 1-aminocyclopropane-1-carboxylic acid oxidase (ACCO) into an enzyme that promotes enantioselective C(sp3)–H fluorinations (example product pictured). Directed evolution is based on cycles of mutations and screening/selection—mimicking natural evolution, but at much higher speeds and aiming to optimize the enzyme for a specific application.
The optimized C–H fluorinating enzyme showed a catalytic activity higher than that of the natural enzyme by a factor of 200. It was used to convert a range of substrates into organofluorine products with high enantioselectivities, and allows the preparation of such enantioenriched organofluorides on a gram scale.
The team found that the most useful mutations in the engineered enzyme were not at the active, iron-containing site, but instead part of the substrate tunnel that allows the substrate to reach the active site within the enzyme. According to the researchers, the evolved Fe-dependent enzyme represents the first new-to-nature C–F bond-forming biocatalyst.
- Biocatalytic enantioselective C(sp3)–H fluorination enabled by directed evolution of nonheme Fe enzymes,
Yang Yang, Liu-Peng Zhao, Binh Khanh Mai, Lida Cheng, Fangqiu Gao, Yunlong Zhao, Rui Guo, Hao Wu, Yongda Zhang, Peng Liu,
ChemRxiv 2024.
https://doi.org/10.26434/chemrxiv-2024-pt58mThis research has been published as a preprint and has not yet been peer-reviewed.
![]()