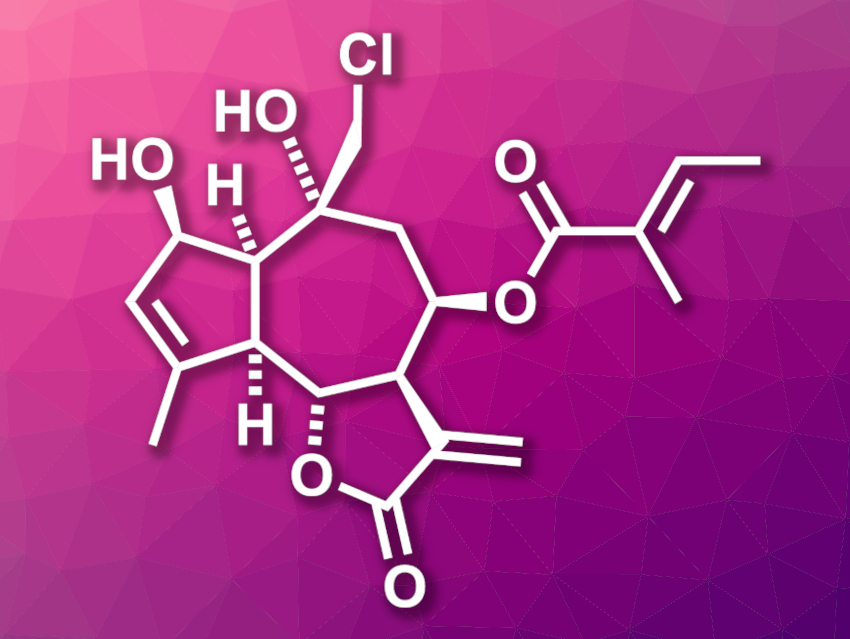
Eupalinilide E (pictured) was isolated from the plant Eupatorium lindleyanum. The compound shows cytotoxic activity against a particular type of lung cancer that is resistant to traditional chemotherapeutic agents. It also could promote the production of human hematopoietic stem and progenitor cells (HSPCs), which could be useful for the treatment of blood diseases. However, natural sources of eupalinilide E are scarce.
Ramkrishna Maity and Saumen Hajra, Centre of Biomedical Research (CBMR), Lucknow, India, have developed a concise and scalable asymmetric total synthesis of eupalinilde E. The team started from (R)-(−)-carvone, which was first converted to a chlorohydrin and then to a O-tosylchlorohydrin. In a key step, a tandem Favorskii rearrangement–elimination to give a cyclopentene carboxylic acid intermediate. This intermediate was converted to the corresponding aldehyde.
The second building block, an allylboronate, was prepared from an O-protected propargyl alcohol. The two synthesized building blocks were combined using a tandem allylboration–lactonization, closing the lactone ring of the target product. The core of the compound was completed using a sequential one-pot oxidation and ene-cyclization. Four more functionalization steps were then needed to obtain the desired product.
Overall, eupalinilide E was synthesized in twelve steps and 20 % yield. The approach involves only six chromatographic purifications. It is scalable and should allow the synthesis of eupalinilde E analogues.
- Asymmetric Total Synthesis of Eupalinilide E, a Promoter of Human HSPC Expansion,
Ramkrishna Maity, Saumen Hajra,
Org. Lett. 2022.
https://doi.org/10.1021/acs.orglett.2c01684
Update (July 7, 2002)
An earlier version of the article had an error in the researcher affiliations. This has been corrected.
![]()