Computational methods have become important for small molecule drug discovery. However, the approaches used there can also be extended to larger systems. Jean-Louis Reymond, University of Bern, Switzerland, and colleagues have, for the first time, applied computational methods to a new class of compounds, known as antimicrobial peptide dendrimers (AMPD), which show promise for combatting multidrug-resistant bacterial strains.
The team built a compound library containing 50 625 different AMPDs by permutation of the peptides’ amino acid sequences. Each chemical structure was encoded as a series of numbers, called fingerprints, which are easily processed by a computer. The machine then created a “map” of different drug candidates in a virtual chemical space, with similar compounds close together. The researchers can use the map to guide their research by focusing on the most promising compounds, the so-called nearest neighbors of known active members of the virtual library. This approach allows the virtual screening of a large number of compounds without the huge effort that would be required to evaluate all candidates experimentally.
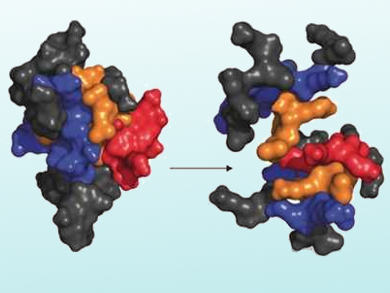
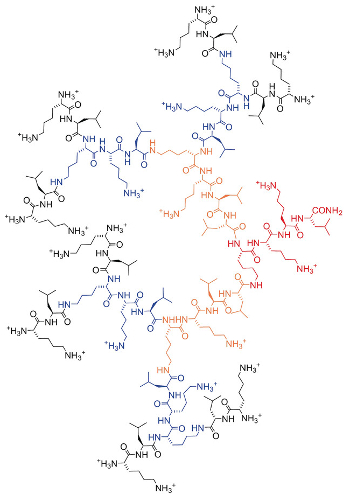
The researchers selected, synthesized, and tested a small number of compounds among the nearest neighbors of a known active AMPD. They obtained several vastly improved drug candidates. The best AMPD, dubbed T7 (pictured below), showed an extended activity spectrum against Gram-negative multidrug-resistant bacteria, high serum stability, and promising in vivo activity.
- Optimizing Antimicrobial Peptide Dendrimers in Chemical Space,
Thissa N. Siriwardena, Alice Capecchi, Bee-Ha Gan, Xian Jin, Runze He, Dengwen Wei, Lan Ma, Thilo Köhler, Christian van Delden, Sacha Javor, Jean-Louis Reymond,
Angew. Chem. Int. Ed. 2018, 57, 8483–8487.
https://doi.org/10.1002/anie.201802837



