Catalytic reactions for the formation of bonds to carbon have been extensively investigated, with impressive results both in activity and selectivity. As a result, many fundamental transformations are well established and provide a synthetic toolkit for the organic chemist. In sharp contrast, analogous processes for the formation of bonds between heavier main group elements (for example phosphorus, silicon, nitrogen, and boron) have not been as extensively investigated and the field remains in its infancy [1].
The desire to develop efficient routes towards inorganic materials is encouraged in part by the fact that they have shown remarkable promise over a wide range of applications, from functional components of biomedical equipment to ceramic thin films, which are present in many electronic devices [2,3]. In particular, the possibility of synthesizing inorganic polymers and inorganic-organic hybrid polymers is highly attractive, as this would allow the development of new materials with potentially interesting chemical and physical properties.
The wide range of oxidation states, coordination numbers, and structures accessible to heavier main group elements presents a wealth of opportunities that is unattainable with purely organic materials. In addition, compared with C–C bonds, heavier main group E–E bonds (for example, Si–O or P–N bonds) can be more oxidatively stable and more conformationally flexible. This could provide polymers with useful properties such as low-temperature flexibility and inherent flame retardancy [4–6].
Despite the fact that main group molecules hold great potential for a number of applications, the general lack of sophisticated routes towards such materials has hindered advancement. Crucially, the synthetic protocols that are so successful for organic chemistry are difficult or impossible to apply to inorganic chemistry [7].
Why Are They Difficult to Make?
If we consider the two main mechanisms available in the synthesis of hydrocarbon polymers (that is, chain-growth and step-growth mechanisms), we can begin to understand the challenges.
Chain-Growth Polymerization
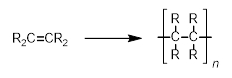
Firstly, chain-growth polymerization involves the sequential addition of each monomer to a growing chain and often, in organic chemistry, relies on the presence of unsaturated bonds which can be broken and used to react with other monomer units (Scheme 1).
 |
|
Scheme 1. Chain-growth polymerization for the formation of hydrocarbon polymers. |
However, with regard to heavier main group chemistry (i.e., that of the third period and lower), there is a distinct lack of analogous monomers with unsaturated bonds, as the synthesis of such molecules is not trivial. The difficulty is primarily due to the weaker bonds formed between the heavier main group elements as a result of increasingly diffuse orbitals and, therefore, less efficient orbital overlap.
Consequently, when attempting to prepare multiply bonded species with heavier main group elements in the past, researchers isolated only singly bonded, cyclic species, which are not useful for chain-growth polymerization (Scheme 2). This observation led to the introduction of the “double bond rule”, stating that the synthesis of multiply bonded species was not possible for elements of the third period and lower [8].
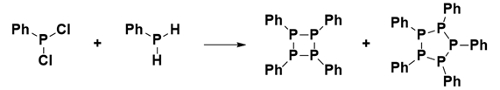 |
|
Scheme 2. Attempted synthesis of a P=P double bond resulting in cyclic oligomers. |
Later, the rule was disproven when it was discovered that sterically bulky groups placed on the main group element could be used to prevent the formation of rings and produce the desired E=E double bonds [9]. However, the presence of such bulky groups typically precludes the molecules’ use in polymerization due to the considerable steric strain that would result (Scheme 3).
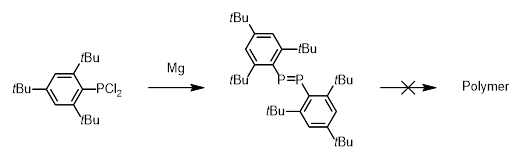 |
|
Scheme 3. Steric protection of diphosphenes which precludes their use in polymerization. |
It should be noted, however, that some examples of living-chain polymerization, i.e., polymerization in which chain termination is not possible, have been successfully used for inorganic substrates. A rare example of this is the polycondensation of N-silylphosphoranimine (Cl3P=NSiMe3) to polyphosphazene [N=PR2]n, reported in 1995 [10].
Step-Growth Polymerization
But what about step-growth polymerization? In organic chemistry, this polymerization mechanism requires two functional groups to be present on either end of a single monomer (denoted X and Y in Scheme 4). These functional groups can react with a complementary functional group on a second monomer, joining the two together and eliminating a small molecule in the process.
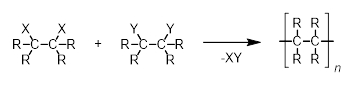 |
|
Scheme 4. Step-growth polymerization for the formation of hydrocarbon polymers. |
However, difunctional monomers based on inorganic species are typically highly reactive and, therefore, are difficult to synthesize and purify [11]. This problem is further exacerbated by the lack of sophisticated routes towards main group molecules. Until quite recently, researchers have been confined to outdated reactions relying on chemically harsh reagents.
Overcoming the Challenges
Dehydrocoupling
Nevertheless, recent advances in the field have been realized, with catalytic dehydrogenative coupling (dehydrocoupling) emerging as a viable strategy [1,12,13]. Broadly speaking, dehydrocoupling is defined as the generation of a new chemical bond between elements (E) with an elimination of hydrogen at the same time. The main advantage of this strategy lies in the far milder reaction conditions, which tolerate a wider variety of substrates and allow greater control over product selectivity.
E–H + H–E’ → E–E’ + H2
It is important to note that dehydrocoupling typically requires the use of a catalyst to promote the reaction. A considerable proportion of the pioneering work in catalytic dehydrocoupling had examined catalysts based on either titanium or zirconium. These catalysts are often preferred because they have a demonstrably superior reactivity and, relatively speaking, a low cost.
In 1985, Harrod and co-workers [14] demonstrated the use of these titanium- or zirconium-based catalysts in the synthesis of polysilanes. Polysilanes are incredibly interesting materials due to their unusual optoelectronic properties, which make them suitable for the fabrication of light emitting diodes (LEDs) for flat-panel displays. This dehydrogenative strategy resulted in greater reproducibility and better control over the reaction, and it could be applied to a wider range of substrates than traditional methods.
One particularly significant piece of work was reported by Stephan et al. in 1995 [15], in which researchers demonstrated the use of a zirconium catalyst to form the unique P16 macrocycle, the largest phosphorus macrocycle synthesized to date [16]. This research has highlighted the potential of catalytic dehydrocoupling to produce new, previously inaccessible materials.
Polycondensation
In addition to dehydrocoupling, another emerging strategy in the synthesis of main group materials is polycondensation with the elimination of a halosilane. This route has been used for the synthesis of heavier main group analogues of poly(p-phenylenevinylene)s (PPVs, Scheme 5), which are π-conjugated polymers. Organic PPVs are interesting in themselves and have a number of applications in LEDs and photovoltaic devices. However, the inclusion of heavier main group elements, which possess lone pairs of electrons that could potentially participate in π-conjugation, could allow access to significantly altered electronic properties.
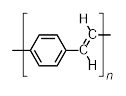 |
|
Scheme 5. General structure of an organic PPV. |
A particularly noteworthy use of this polycondensation synthetic approach is in the preparation of the first reported π-conjugated polymer containing P=C bonds, as reported by Wright and Gates in 2002 (Scheme 6) [17]. Pure product was obtained in a 35 % yield with a moderate degree of polymerization across batches. Preliminary studies using UV/Vis spectroscopy revealed a shift in the absorbance spectrum to a lower frequency compared with representative model oligomers. This suggests a degree of π-conjugation through the phenylene and P=C units.
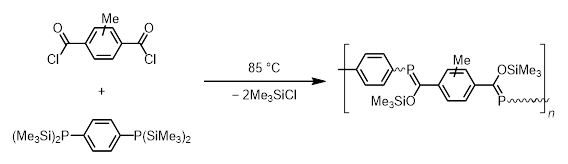 |
|
Scheme 6. Preparation of polymer containing P=C bonds [17]. |
Sterically Demanding Organic Spacers
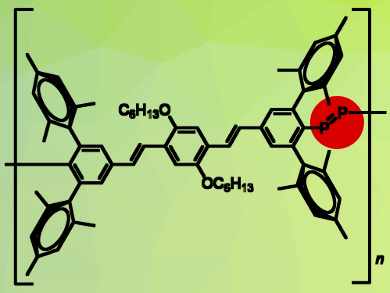
Another method for the synthesis of inorganic/organic hybrid PPVs is the use of sterically demanding organic spacers [11]. These spacers have been carefully designed so that they can provide the necessary steric protection to support two heavier main group double bonds, without preventing polymerization. In 2004, Smith and Protasiewicz [18] reported a bifunctionalized ligand with a sterically demanding organic spacer that can be polymerized to the respective diphosphene-PPV (Scheme 7). The polymer, which contains P=P double bonds, was obtained in near-quantitative yields by heating neat samples of the diphospha-Wittig ligand to 250 °C for just two minutes. Significantly, this was the first reported synthesis of a polymer containing a multiple bond between two heavier main group species within the polymer backbone.
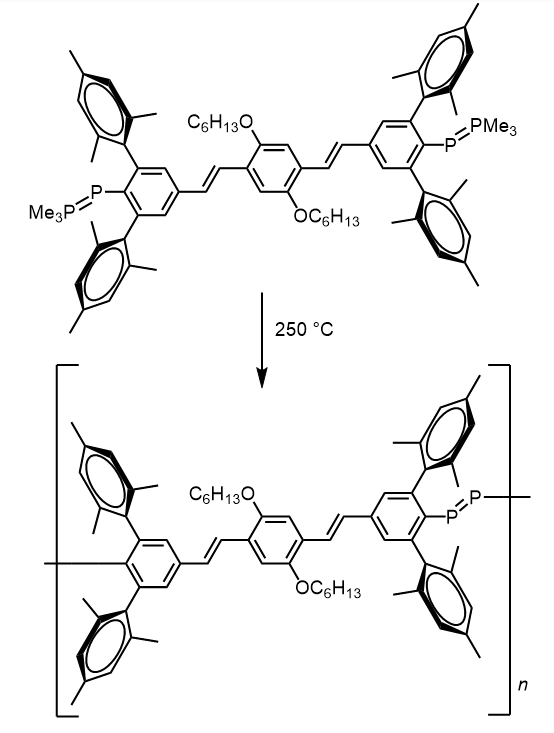 |
|
Scheme 7. Synthesis of diphosphene-PPV using sterically demanding organic spacer. |
Where We Are Today
Over the past few decades, there has been increasing interest in the synthetic chemistry of heavy main group elements, with many research groups now making this their main focus.
Catalytic dehydrocoupling of amine-boranes (B–N coupling) has become a particularly popular topic of research, as they are ideal candidates for hydrogen storage and could be key to advancing the hydrogen economy. The Waterman group [19] reported the use of a sophisticated zirconium-based catalyst which operated effectively at moderate temperature and low to moderate catalyst loadings. Interestingly, they were able to use this methodology for the hydrogenation of alkenes using amine boranes as the sacrificial hydrogen source.
Furthermore, a recent breakthrough reported by the Manners group [20] demonstrated the first titanium-catalyzed dehydropolymerization of amine-boranes to high-molar-mass polymers. Previous synthetic routes not only required the use of rare earth metal catalysts, but were also significantly more limited in their synthetic scope.
A further development in the field of B–N coupling was reported by the Hill group. The method makes use of an alkali-metal catalyst based on magnesium and calcium [21]. In addition, the Wright group demonstrated the use of a tin-based catalyst for the catalytic dehydrocoupling of phosphines [22]. Since catalysis has been, until recently, mainly restricted to expensive, rare-earth-metal complexes, these developments in main group catalysis are highly promising [23].
With regards to polycondensation methods, recent progress has seen the discovery of the boron-nitrogen analogue of PPV as well as novel linear poly(iminoborane)s, which are currently being investigated for applications in materials science [24,25].
It is clear that much progress in the field of catalytic dehydrocoupling has been made over recent years. The development of chemically milder synthetic strategies has resulted in a variety of interesting molecular species and polymers with applications spanning across all areas of science. However, synthetic inorganic chemistry remains comparatively far behind organic chemistry and there is still a large synthetic scope to be realized in the future.
References
[1] E. M. Leitao et al., Catalysis in service of main group chemistry offers a versatile approach to p-block molecules and materials, Nat. Chem. 2013, 5, 817–829. https://doi.org/10.1038/nchem.1749
[2] A. K. Franz , S. O. Wilson, Organosilicon Molecules with Medicinal Applications, J. Med. Chem. 2013, 56, 388–405. https://doi.org/10.1021/jm3010114
[3] T. Wideman, L. G. Sneddon, Dipentylamine-Modified Polyborazylene: A New, Melt-Spinnable Polymeric Precursor to Boron Nitride Ceramic Fibers, Chem. Mater. 1996, 8, 3–5. https://doi.org/10.1021/cm950398b
[4] J. E. Mark et al., Inorganic Polymers, Oxford University Press, Inc., New York, 2nd Ed., 2005. ISBN: 9780195131192
[5] I. Manners, Polymer science with transition metals and main group elements: Towards functional, supramolecular inorganic polymeric materials, J. Polym. Sci. Part A Polym. Chem. 2002, 40, 179–191. https://doi.org/10.1002/pola.10069
[6] S. Rothemund, I. Teasdale, Preparation of polyphosphazenes: a tutorial review, Chem. Soc. Rev. 2016, 45, 5200–5215. https://doi.org/10.1039/c6cs00340k
[7] I. Manners, Polymers and the Periodic Table: Recent Developments in Inorganic Polymer Science, Angew. Chem. Int. Ed. Engl. 1996, 35, 1602–1621. https://doi.org/10.1002/anie.199616021
[8] H. Kӧhler, A. Michaelis, Ueber Phenylphosphin und Phosphobenzol (Diphosphenyl) (in German), Ber. Dtsch. Chem. Ges. 1877, 10, 807–814. https://doi.org/10.1002/cber.187701001222
[9] M. Yoshifuji et al., Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene: isolation of a true phosphobenzene, J. Am. Chem. Soc. 1981, 103, 4587–4589. https://10.1021/ja00405a054
[10] C. H. Honeyman et al., Ambient Temperature Synthesis of Poly(dichlorophosphazene) with Molecular Weight Control, J. Am. Chem. Soc. 1995, 117, 7035–7036. https://doi.org/10.1021/ja00131a040
[11] A. M. Priegert et al., Polymers and the p-block elements, Chem. Soc. Rev. 2016, 45, 922–953. https://doi.org/10.1039/c5cs00725a
[12] S. Greenberg, D. W. Stephan, Stoichiometric and catalytic activation of P–H and P–P bonds, Chem. Soc. Rev. 2008, 37, 1482–1489. https://doi.org/10.1039/b612306f
[13] R. Waterman, Mechanisms of metal-catalyzed dehydrocoupling reactions, Chem. Soc. Rev. 2013, 42, 5629–5641. https://doi.org/10.1039/c3cs60082c
[14] C. Aitken et al., Polymerization of primary silanes to linear polysilanes catalyzed by titanocene derivatives, J. Organomet. Chem. 1985, 279, C11-13. https://doi.org/10.1016/0022-328X(85)87029-7
[15] M. C. Fermin, D. W. Stephan, Catalytic Oligomerization of Primary Phosphines by the Anionic Zirconocene Trihydride: [Cp*2ZrH3]–, J. Am. Chem. Soc. 1995, 117, 12645–12646. https://doi.org/10.1021/ja00155a033
[16] N. Etkin et al., Catalytic Synthesis of the P16 Macrocycle (C6H4P2)8, J. Am. Chem. Soc. 1997, 119, 2954–2955. https://doi.org/10.1021/ja964203s
[17] V. A. Wright, D. P. Gates, 3.0.CO;2-6″ target=”_blank”>https://doi.org/10.1002/1521-3773(20020703)41:13<2389::AID-ANIE2389>3.0.CO;2-6
[18] R. C. Smith, J. D. Protasiewicz, Conjugated Polymers Featuring Heavier Main Group Element Multiple Bonds: A Diphosphene-PPV, J. Am. Chem. Soc. 2004, 126, 2268–2269. https://doi.org/10.1021/ja0394683
[19] K. A. Erickson et al., Zirconium-Catalyzed Amine Borane Dehydrocoupling and Transfer Hydrogenation, Organometallics 2015, 34, 4693–4699. https://doi.org/10.1021/acs.organomet.5b00415
[20] T. Jurca et al., Step-growth titanium-catalysed dehydropolymerisation of amine–boranes, Chem. Sci. 2018, 9, 3360–3366. https://doi.org/10.1039/c7sc05395a
[21] D. J. Liptrot et al., Alkaline‐Earth‐Catalyzed Dehydrocoupling of Amines and Boranes, Angew. Chem., Int. Ed. 2015, 54, 13362–13365. https://doi.org/10.1002/anie.201505949
[22] V. Naseri et al., Stoichiometric and catalytic Sn-mediated dehydrocoupling of primary phosphines, Chem. Commun. 2010, 46, 5000–5002. https://doi.org/10.1039/c0cc00827c
[23] P. P. Power, Main-group elements as transition metals, Nature 2010, 463, 171–177. https://doi.org/10.1038/nature08634
[24] T. Lorenz et al., Poly(p‐phenylene iminoborane): A Boron–Nitrogen Analogue of Poly(p‐phenylene vinylene), Angew. Chem., Int. Ed. 2017, 56, 2780–2784. https://doi.org/10.1002/anie.201612476
[25] O. Ayhan et al., Cyclolinear Oligo‐ and Poly(iminoborane)s: The Missing Link in Inorganic Main‐Group Macromolecular Chemistry, Chem. Eur. J. 2018, 24, 5883–5894. https://doi.org/10.1002/chem.201705913
Author Information
Bethany Lawson
University of St. Andrews, UK