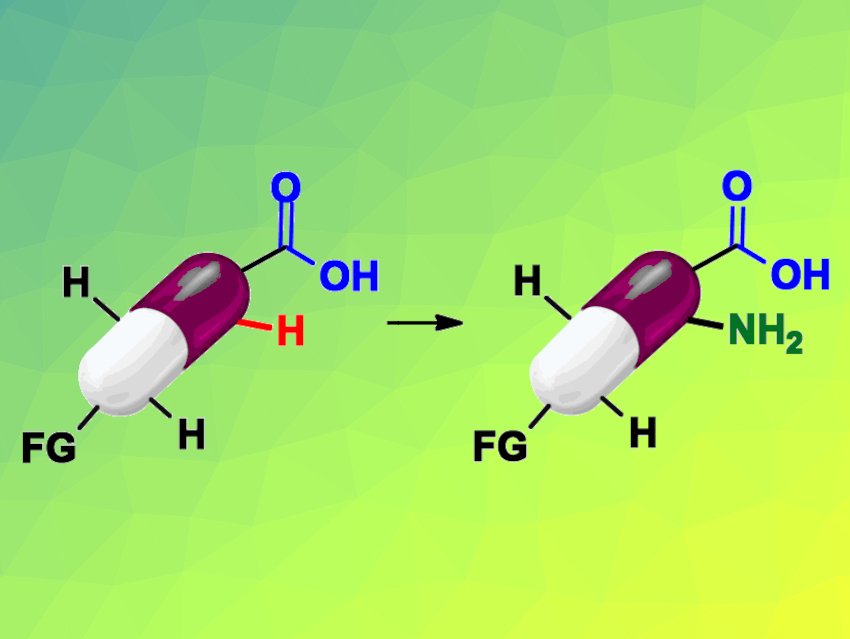
C–H bonds are ubiquitous in pharmaceuticals, and C–H functionalizations are useful in drug discovery. However, limitations in functional-group tolerance and substrate scope can hinder the use of such reactions, especially for late-stage functionalizations. Benzoic acids, for example, frequently occur in drugs and compounds relevant for drug development.
Magnus J. Johansson and Erik Weis, Stockholm University, Sweden, and AstraZeneca, Mölndal, Sweden, and Belén Martín-Matute, Stockholm University, have developed a mild, iridium-catalyzed, ortho-selective C−H amination of benzoic acids (pictured schematically). The team used Cp*Ir(H2O)3SO4 as a catalyst, p-toluenesulfonyl azide (TsN3) as a nitrogen source, Et3N as a base, and 1,1,1,3,3,3-Hexafluoroisopropanol (HFIP) as the solvent.
Using this approach, the researchers were able to functionalize a variety of benzoic acids and perform late-stage functionalizations of different marketed drugs. The method allows not only the synthesis of high-value drug analogues, but also the introduction of linkers designed for conjugation chemistry. The latter has so far been mostly unexplored in terms of C–H activation chemistry and has great potential for drug-discovery applications. The developed approach can also be used in high-throughput experimentation.
- Late‐stage amination of drug‐like benzoic acids: Access to anilines and drug conjugates via directed iridium‐catalyzed C−H activation,
Belén Martín-Matute, Erik Weis, Magnus J. Johansson,
Chem. Eur. J. 2021.
https://doi.org/10.1002/chem.202103510
![]()