Addressing issues of chemo-, regio-, diastereo-, and enantioselectivity is of utmost importance for synthetic chemistry, with great effort being made to develop highly selective reactions. Understanding the origins and mechanisms of these experimentally observed, often puzzling, selectivities is also beneficial for the enhancement of synthetic methodology. A great strength of computational approaches, particularly electronic structure theory, is the ability to unravel puzzles of this type through the elucidation of detailed reaction mechanisms.
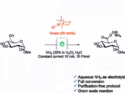
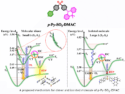
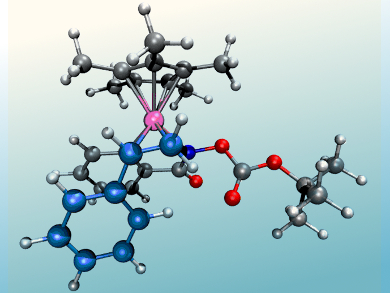
Clémence Corminboeuf, Nicolai Cramer, and colleagues, Ecole Polytechnique Fédérale de Lausanne (EPFL), Switzerland, have used a computational approach to understand and take control of the regioselectivity of RhIII-catalyzed directed C–H functionalizations of arylhydroxamates. Two different catalytic systems were developed, one utilizing the 1,2-disubstituted cyclopentadienyl ligand CpCy and the other Cp*, that allow the selective formation of regioisomeric 3-aryl dihydroisoquinolones and previously inaccessible 4-aryl dihydroisoquinolones, respectively, under full catalyst control.
The differences in the catalysts were computationally examined using density functional and transition-state theory, calculating different possible pathways to elucidate key contributing factors leading to the regioisomeric products. These factors were identified as the stabilities of the initially formed rhodium complex styrene adducts and activation barrier differences for the migratory insertion.
- Ligand-Controlled Regiodivergent Pathways of Rhodium(III)-Catalyzed Dihydroisoquinolone Synthesis: Experimental and Computational Studies of Different Cyclopentadienyl Ligands,
Matthew D. Wodrich, Baihua Ye, Jérôme F. Gonthier, Clémence Corminboeuf, Nicolai Cramer,
Chem. Eur. J. 2014.
DOI: 10.1002/chem.201404515