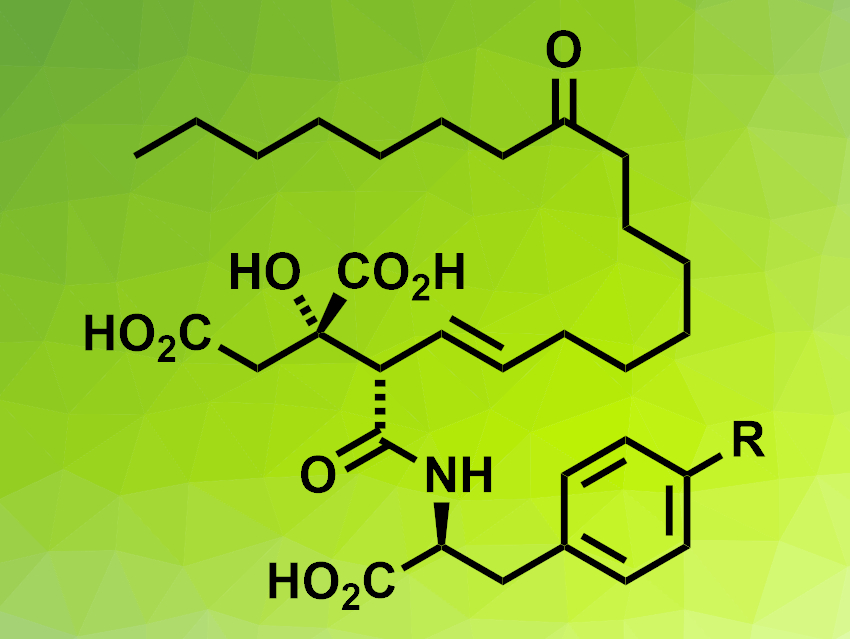
Viridiofungins A and B (pictured, R = OH for viridiofungin A, R = H for viridiofungin B) are part of a family of bioactive alkyl citrate natural products and were first isolated from a fungus. The compounds feature a fairly long alkyl chain and a citric acid unit and can be challenging to synthesize in a stereoselective manner. Existing syntheses of viridiofungin A have 16 or more steps [1].
Mark A. Rizzacasa, The University of Melbourne, Victoria, Australia, and colleagues have developed a highly stereoselective total synthesis of viridiofungins A and B via a common β-lactone intermediate in 13 steps. The team started from commercially available (S)-(+)-γ-hydroxymethyl-γ-butyrolactone, which was protected and then subjected to a formal [2+2] cycloaddition with di-t-butylacetylene dicarboxylate. The resulting cyclobutene diester underwent an HF-mediated rearrangement to give a bicyclic lactone. This lactone was converted to the desired common alkene-functionalized β-lactone intermediate via a ring-opening, a lactonization, and an alkene isomerization.
The team subjected this intermediate to an olefin cross-metathesis reaction to introduce the side chain of the target compounds. Then, a ring-opening of the β-lactone was used to form the amide unit and introduce the correct aromatic substituent for viridiofungin A or B, and a final deprotection step gave the desired products in high yields. According to the researchers, the stable β-lactone intermediate could also be useful as a precursor for other viridiofungins or their analogues for further studies.
- Total Synthesis of Viridiofungins A and B,
Liselle Atkin, Angus Robertson, Jonathan M. White, Mark A. Rizzacasa,
Org. Lett. 2021.
https://doi.org/10.1021/acs.orglett.1c00971
Reference
- [1] Synthetic Studies of Viridiofungins, Broad-Spectrum Antifungal Agents and Serine Palmitoyl Transferase Inhibitors,
Naoya Kumagai, Masakatsu Shibasaki,
J. Antibiot. 2017, 71, 53–59.
https://doi.org/10.1038/ja.2017.110
![]()