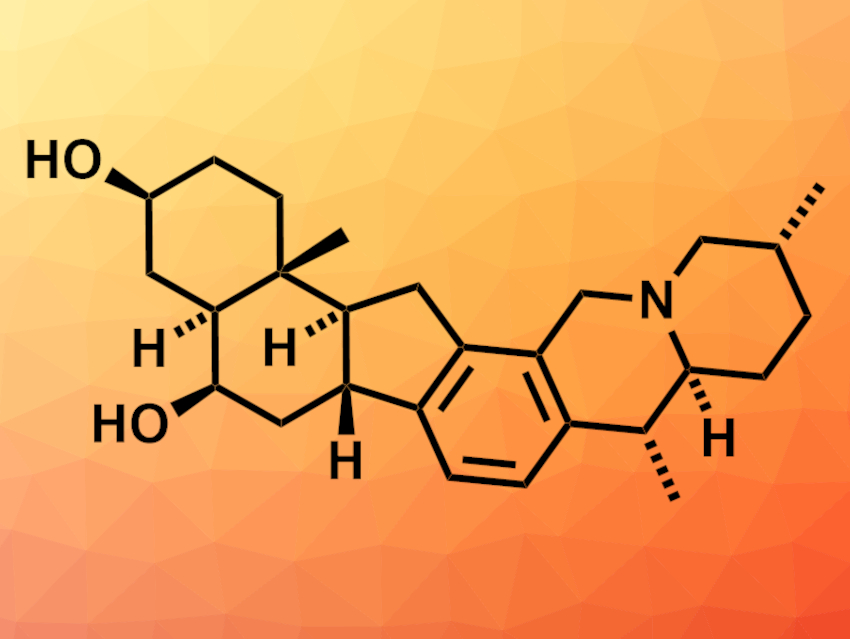
Alkaloid natural products can have a variety of biological activities. Heilonine (pictured), for example, is a hexacyclic alkaloid with a challenging structure for total synthesis. Known syntheses of similar compounds require a large number of steps.
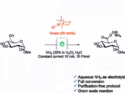
Kyle J. Cassaidy and Viresh H. Rawal, University of Chicago, IL, USA, have performed a convergent enantioselective total synthesis of (+)-heilonine. The team’s strategy involves a transition-metal-catalyzed [2+2+2] cycloisomerization reaction, which creates three new C–C bonds in one step. The researchers first enantioselectively synthesized a diyne fragment that contains the A and B rings of the target compound. The fragment was connected to a propargyl-substituted piperidinone that contains the F ring. An intramolecular [2+2+2] cycloisomerization reaction using Wilkinson’s catalyst was then used to close the C, D, and E rings in one step. With the carbon skeleton completed, further functionalization steps gave the desired product.
The team obtained (+)-heilonine in 21 steps (starting from ethyl vinyl ketone). According to the researchers, the convergent, modular approach could also be useful to access related natural compounds and other analogs. This could allow studies of their biological activities.
- Enantioselective Total Synthesis of (+)-Heilonine,
Kyle J. Cassaidy, Viresh H. Rawal,
J. Am. Chem. Soc. 2021.
https://doi.org/10.1021/jacs.1c08756
![]()