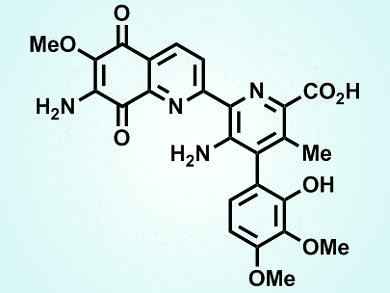
(±)-Streptonigrin (pictured) is a potent broad-spectrum anticancer antibiotic. Although the drug reached phase II clinical trials in the late 1970s, trials were stopped owing to its toxicity. Despite this fact, there is still considerable interest in this area because analogues of streptonigrin may be less toxic but equally effective as anticancer agents.
Timothy Donohoe and colleagues, University of Oxford, UK, and Federal University of Minas Gerais, Brazil, planned a convergent route, in which three fragments are formed before being joined together to create the tetracyclic core of streptonigrin. Two routes were devised and both rely on a ring-closing metathesis reaction to form one of the nitrogen-containing (pyridine) rings, and a challenging (Suzuki−Miyaura) coupling reaction. The shorter of the sequences gave streptonigrin in 14 steps and 11 % overall yield.
A convergent approach is attractive because it provides good opportunities for various analogues to be formed. Each of the three fragments may be derivatized before being joined together. The team hopes this route will facilitate the synthesis of a library of streptonigrin analogues for biological evaluation.
- Total Synthesis of the Antitumor Antibiotic (±)-Streptonigrin: First- and Second-Generation Routes for de Novo Pyridine Formation Using Ring-Closing Metathesis,
Timothy J. Donohoe, Christopher R. Jones, Anne F. Kornahrens, Luiz C. A. Barbosa, Louise J. Walport, Matthew R. Tatton, Michael O’Hagan, Akshat H. Rathi, David B. Baker,
J. Org. Chem. 2013.
DOI: 10.1021/jo402388f