Density functional theory (DFT) is a tried and true method for exploring the electronic properties of materials, but venturing into new territory can produce unexpected results. James Ibers, Northwest University, Evanston, IL, USA, and colleagues found this out when they characterized BaUSe3 during an exploration of the Ba/U/Se ternary system.
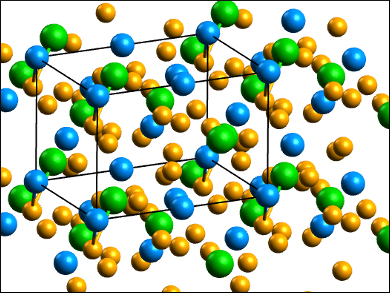
The BaUSe3 crystal structure (pictured) is related to the perovskite structure. Its calculated activation energy (0.12 eV), based on resistivity measurements, is typical of a narrow band gap semiconductor, consistent with the black color of the compound. The UV-vis spectrum confirmed that BaUSe3 has no optical band gap greater than 1 eV. However, DFT calculations based on the crystal structure predict a band gap of 2.5 eV, characteristic of a relatively large band gap semiconductor.
The structural results were obtained at 100 K, and the resistivity measurements and UV-vis spectrum were obtained at 298 K, but BaUSe3 has no phase transition between these two temperatures. This large discrepancy in band gap values was not seen in many previous studies of similar compounds. The researchers suggest a shortcoming in the DFT calculations when dealing with this actinide compound.
- Synthesis, Crystal Structure, Theoretical, and Resistivity Study of BaUSe3,
Jai Prakash, Maria S. Tarasenko, Adel Mesbah, Sébastien Lebègue, Christos D. Malliakas, James A. Ibers,
Inorg. Chem. 2016.
DOI: 10.1021/acs.inorgchem.6b01202



