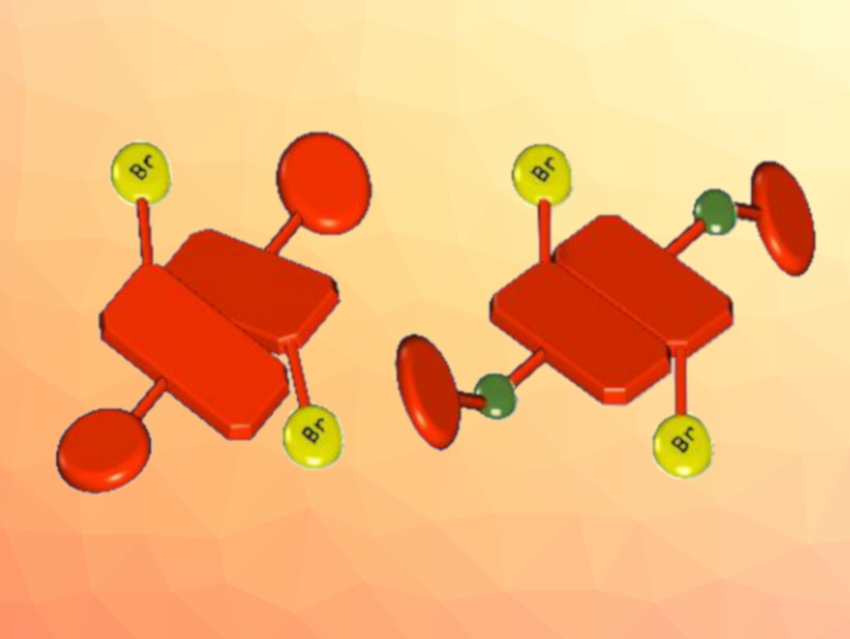
Perylene-diimides (PDI) are widely used in the field of organic electronics due to their useful n-semiconducting properties, chemical and thermal robustness, and ease of functionalization. Bulk solids and thin-films of these species crystallize in a variety of stacking configurations, depending on the conformation of the polyaromatic core. Work on 1,7-dibromoperylene derivatives has been somewhat limited because the heavily twisted nature of the perylene core may prevent the formation of efficient π–π stacks and could hamper the charge mobility.
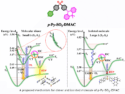
Norberto Masciocchi, University of Insubria, Como, Italy, and colleagues have found that 1,7-dibromosubstituted perylene-diimides (pictured below), which usually have a heavily twisted conformation in the gas phase, in solution, and in the solid state (pictured schematically above on the left), can be easily “flattened” in the solid state (pictured schematically above on the right).
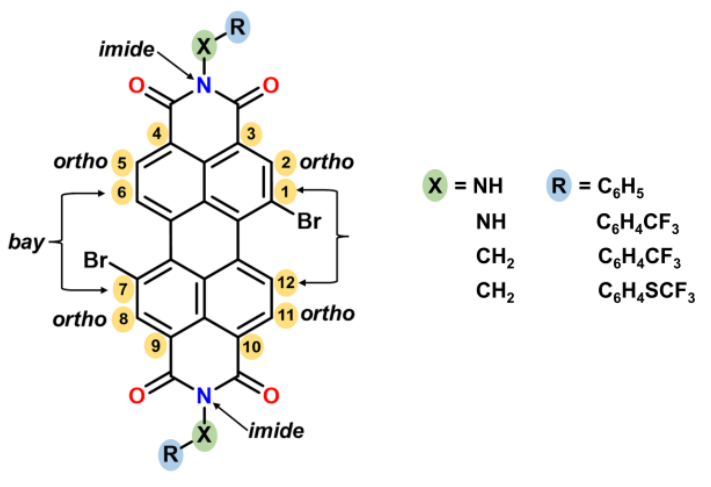
The team used axial imide substituents with low stereochemical hindrance (CH2–R or NH–R) to promote the formation of such stacked, “core-flattened” PDI molecules. The four isolated species were synthesized from 3,4,9,10-tetracarboxylperylene dianhydride, which was brominated and then converted to the desired imides via reactions with phenylhydrazine or benzylamine derivatives.
X-ray powder diffraction and density functional theory (DFT) showed that thanks to the flexibility of the N–X–Ar link (X = CH2, NH), flat cores are obtained in the solid state. This packing is the result of stabilizing intramolecular interactions due to the formation of stacks of flat perylene cores. According to the researchers, this strategy may be useful to induce “anomalously flat” polyaromatic cores and promote suitable crystal packing and orbital interactions for improved electronic performance.
- Forcing Twisted 1,7‐Dibromoperylene Diimides to Flatten in the Solid State: What a Difference an Atom Makes,
Konstantis F. Konidaris, Marco Zambra, Francesco Giannici, Antonietta Guagliardi, Norberto Masciocchi,
Angew. Chem. Int. Ed. 2023.
https://doi.org/10.1002/anie.202310445
![]()