Compounds containing nitrogen–silicon bonds are widely used in various fields of chemistry and materials science. The most common synthesis route for these compounds is the reaction of chlorosilanes with amines, which has a relatively low atom economy due to the formation of stoichiometric amounts of hydrochloric acid as a byproduct. To circumvent this problem, various catalytic processes have been developed. However, they generally lead to the loss of the silicon–hydrogen bond, which can be useful for further reactions.
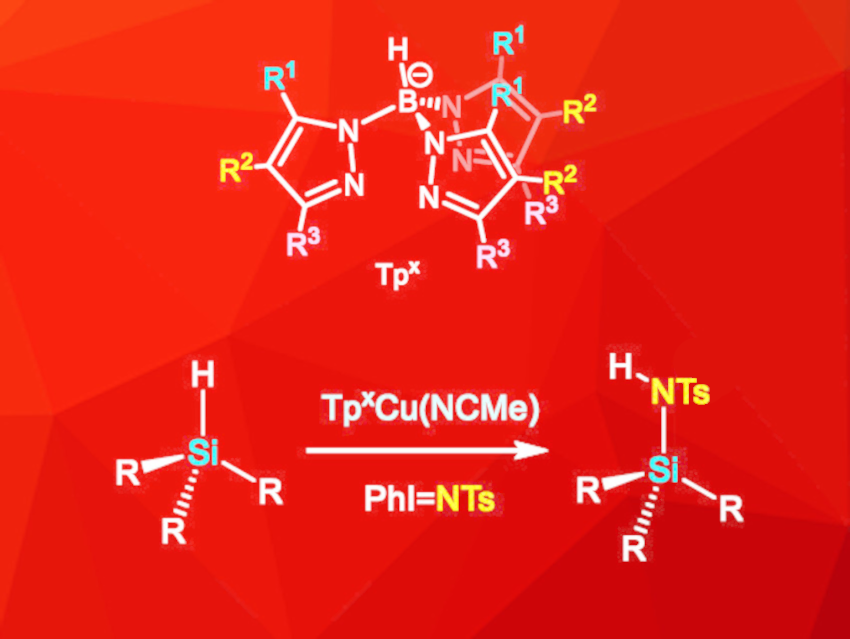
Feliu Maseras, Institute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Tarragona, Spain, M. Mar Díaz-Requejo, Pedro J. Pérez, Universidad de Huelva, Spain, and colleagues have developed a copper-catalyzed amination of silanes via nitrene insertion in which the hydrogen atom is retained (pictured). The reaction is catalyzed by a copper complex with a ligand of the type Tpx (trispyrazolylborate, pictured), and PhI=NTs ([N-(p-toluenesulfonyl)imino]phenyliodinane) was used as the nitrene precursor. The reaction was performed in dichloromethane at room temperature under an inert atmosphere.
A wide range of hydrosilanes with aryl and/or alkyl substituents were functionalized, with yields of up to 90 %. Substituents on aryl substituents did not affect the reaction significantly. Alkyl silanes, however, were less reactive, especially if bulky alkyl groups were present. The synthesis route was successfully extended to disilanes and siloxanes, which gave monoaddition products in moderate to high yields. Insertions of two equivalents of the nitrene species into disilane substrates were not observed, even if a large excess of PhI=NTs was used. Overall, the developed synthesis route is suitable for a wide range of substrates and retains the H atom in the substrate, which improves the atom economy compared to previous methods.
- Introducing the Catalytic Amination of Silanes via Nitrene Insertion,
Anabel M. Rodríguez, Jorge Pérez-Ruíz, Francisco Molina, Ana Poveda, Raúl Pérez-Soto, Feliu Maseras, M. Mar Díaz-Requejo, Pedro J. Pérez,
J. Am. Chem. Soc. 2022.
https://doi.org/10.1021/jacs.2c03739
![]()



