Tibor Szilvási, University of Alabama, Tuscaloosa, USA, and his co-author used a combination of atomistic modeling and machine learning to demonstrate how interactions between silver nanoparticles and the surface they rest on change the shape of the silver nanoparticles under real-world conditions. Their research found differences from traditional nanoparticle models.
What did you do?
We combined electronic structure theory calculations with machine learning to study supported nanoparticles 1 to 5 nm in diameter directly under experimental conditions. Our approach showed quantitative agreement with benchmark microcalorimetric measurements confirming the validity of the simulations.
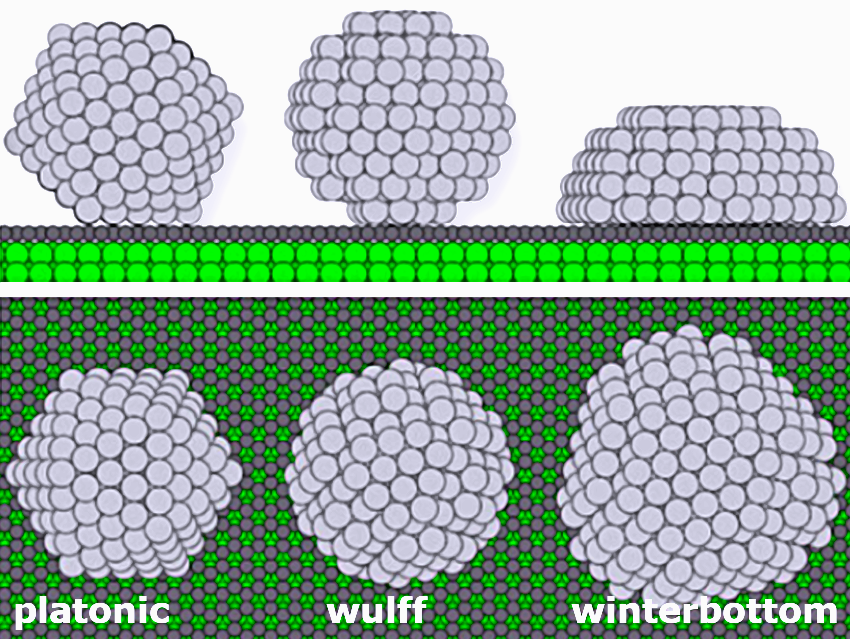
Then, we analyzed the structure of the supported silver nanoparticles and found that their optimal shape does not follow any idealized nanoparticle constructions such as Platonic, Wulff, or Winterbottom. Instead, metal–support interactions reshape the nanoparticle surfaces into a more rounded form.
What is particularly interesting to you about this?
Supported nanoparticles are the workhorse of many catalytic processes, but it is extremely difficult to determine their true atomic structure under working conditions. Experiments like transmission electron microscopy (TEM) or in situ spectroscopy (e.g., XPS, IR) can often miss subtle reconstructions, while traditional simulations (e.g., DFT) rely on oversimplified geometric models.
Our goal was to provide a realistic and validated way to model supported nanoparticles that can bridge experiments and computations.
What is new and cool about this?
We showed that electronic structure theory-quality simulations of supported nanoparticles are now possible at a catalytically relevant nanoparticle size range.
Surprisingly, we found that commonly assumed structural models like the Wulff construction provide inaccurate descriptions for small supported nanoparticles. Instead, optimized nanoparticle structures quantitatively reproduce calorimetry results and reveal reshaping effects where edges and corners smooth out into rounded morphologies.
Our result challenges widely applied assumptions in modeling of nanoparticle catalysis, which are often believed to be questionable, but for which a better approach was previously not viable.
What are your key findings?
- Realistic simulations of supported nanoparticle match state-of-the-art experimental measurements. Optimized structures reproduce adhesion, chemical potential, and heat of adsorption within experimental and computational uncertainties.
- Idealized computational models fail for small supported nanoparticles. Wulff and Platonic constructions miss key structural features at the contact with the support, while the Winterbottom construction performs better but still does not perfectly capture the rounding of nanoparticles.
- Catalytic descriptors change for optimized nanoparticle structures. Coordination numbers, strain distributions, and active site populations shift compared to widely assumed traditional models, affecting how catalytic activity should be predicted in multiscale kinetic modeling.
What is the longer-term vision for your research?
Our methodology opens the door to predictive modeling of supported nanoparticles under catalytic conditions. The long-term vision is to make atomically resolved and computationally efficient nanoparticle simulations standard practice in catalysis and materials science.
Applying machine learning-based methods will enable new frontiers of research where we can bring the computational model up to the size and complexity of the true experimental systems and eliminate many approximations of modeling.
What part of your work was the most challenging?
Training and validating machine learning models for nanoparticles across different sizes were challenging as small nanoparticles have very complex potential energy landscapes, while larger nanoparticles demand efficiency and stability in geometry optimization. This required us to experiment with new approaches, develop our own optimization method, and use an iterative data refinement workflow to find the critical data to add to our machine learning model.
What is the goal of combining computational and experimental approaches in nanoparticle research?
We hope our results bring experiments of nanoparticles under catalytic conditions together with simulations of the same nanoparticles in identical conditions, enabling computations to function similar to in situ spectroscopy. In the future, this approach can help to understand what is occurring on the surface of nanoparticles in a real catalyst and make direct comparisons with a range of spectroscopy and microscopy measurements to provide a holistic view of nanoparticle catalysis.
Thank you very much for sharing these insights.
The paper they talked about:
- Metal-Support Interactions Reshape Nanoparticle Catalyst Surfaces,
Tristan Maxson, Tibor Szilvási,
Angewandte Chemie Novit 2025.
https://doi.org/10.1002/anov.70008

Tibor Szilvási is an Assistant Professor in the Department of Chemical and Biological Engineering at the University of Alabama, Tuscaloosa, USA.
.
Also of Interest


