
Dr. Aleksandar Kondinski works on knowledge engineering in chemistry and is involved in the development of agents that can rationally design new self-assembling materials based on available knowledge and evidence-based reasoning.
Dr. Xiaochi Zhou has been applying natural language processing (NLP) to chemistry even before ChatGPT became popular. He has integrated NLP with knowledge graphs to ensure that the answer given by the machine is explainable. These systems can also be used to break down tasks or perform calculations. Both are based at the University of Cambridge, UK, and at CREATE (Campus for Research Excellence and Technological Enterprise) in Singapore.
Here they talk with Dr. Vera Koester for ChemistryViews about Marie, a system that reliably answers complex chemistry questions, OntoMOPs (Ontology for Metal Organic Polyhedra), a tool that explains and predicts (crystalline) porous materials and supramolecular systems, and future uses of AI in chemistry.
In a nutshell, what are you working on?
Aleksandar Kondinski: We are working on systems that can solve complex chemical problems more efficiently and effectively than conventional methods. Our software agents mimic the decision-making processes of a human expert to perform complex decision-making tasks. This improves the quality of decisions while reducing the time required to solve complex problems.
Talking Chemistry with Computers
You work in a highly interdisciplinary manner, not only within the field of chemistry but also across diverse areas such as chemistry, computer science, and electrical engineering. How does this interdisciplinary approach impact your work?
Xiaochi Zhou: I don’t really have a strong background in chemistry. When I first got the task of constructing a chemistry knowledge graph and implementing a human interface on top of it to retrieve data and get new information from the chemistry data, it was a really big challenge. To be honest, generating a chemistry knowledge graph has a lot in common with generating other knowledge graphs; it has roughly the same structure. So, if you just treat it as a knowledge graph with some different features, then to some extent I don’t have to understand chemistry too much. Instead, I treat it as a different distribution of data, a different structure of data.
But still, domain expertise is important, and I have learned some concepts and ideas about chemistry. For example, I have been working on Aleks’ OntoMOPs, an ontology that allows a detailed semantic description of metal-organic polyhedra (MOPs). A detailed semantic description of a polymer would involve breaking down the term “polymer” into its main components and understanding their meaning and properties. So, before I start building an ontology, I need to understand what each term means. But to be honest, for most of my work, I don’t need to understand the chemistry in too much detail. I just treat it as data, like I said.
Aleksandar Kondinski: How we make decisions and how we keep knowledge always goes back to what I would call a “first principle”. When making decisions or trying to recall information, representing it in a formal way allows for reuse across multiple disciplines [1].
In our group, we deal with chemistry but also chemical engineering (see interview with Professor Kraft). We have colleagues here in the office who deal with buildings, with civil engineering. Of course, domain expertise will always be very important in the way you make decisions. But I think, if you look at the structure and the tools, they are very similar across the different disciplines; they all use similar approaches. This leads to questions like whether chemists should invent a new language to communicate with information systems or rely on existing tools.
What is your opinion?
Xiaochi Zhou: Of course, when we model chemical data and chemical knowledge, we want it to be as interoperable as possible. Interoperability is a very important property for all kinds of data structures because you might not only focus on the domain of chemistry, but you might also have cross-domain implications. At some point, we might want to integrate mathematical data, geographical data, and so on. So, you want the data from different domains to be as similar as possible. That is why we were adopting the knowledge graph technology. It links data and knowledge from different domains and creates a common database for all domains.
Aleksandar Kondinski: Knowledge engineering in chemistry stems from the development of expert systems that mimic the thought processes of chemists. If you go back to the beginning of chemistry, there was no language and structure. Chemists would argue about what language and symbols to use for gold or sulfur and so on. Then, they started discussing how to write reactions, how to write compounds, … It was only in the last, I would say, 50 or 60 years that we started discussing how to write chemistry that is done by a machine. That actually brings us to cheminformatics.
I think it is also a question of whether it is always important for the computer to use the same language as we do, or whether the computer can perform some operations and give us information back in the language we speak.
There are some pioneers like you working in this field, but is there someone who sets the rules for what should be used and how, like IUPAC does for naming molecules and elements?
Aleksandar Kondinski: There are ISO standards for ontologies. However, it often takes years to arrive at a kind of knowledge map and agree on a standard in committees. Sometimes you do not have five or ten years or so to discuss how best to represent a knowledge map. I understand that it is important to have a standard, and so we always agree on the representation of ontologies, for example.
Often, you propose an ontology for the problem you want to solve and what you want to derive from that knowledge. For example, we proposed this ontology for MOPs, OntoMOPs. However, the question is, how do you know if this is the best ontology? Comparable to earlier developments in chemistry, as just mentioned, you need some flexibility to formally structure the knowledge, and, at the same time, the community has to slowly accept or reject whether something is reasonable or unreasonable.
When you are at the forefront of research, I think it is very difficult to have a standard ontology in advance because you are solving a new problem. If you think about the chemists of the 19th century, they often struggled in expressing their understanding of chemistry, by this struggle, advanced the field. I think this is similar to engineering scientists today, who question ideas that already exist and wonder if this is really the best way to understand.
So how are you looking at what is the best way to represent knowledge?
Aleksandar Kondinski: For me, coming from a chemistry background, there is beauty I see in the presentation of knowledge. Maybe that is a bit romantic.
For example, people first described MOPs as shapes. Then I came up with the idea of using assembly models and generic building units to describe MOPs, which was inspired by commercially available interlocking disks for children to construct structures [2,3]. We proposed an ontology based on this idea, the OntoMOPs ontology. Originally, they are called polyhedral because the scientists were looking at their shapes, not how they are constructed. We actually distilled the essentials.
I think it is a positive aspect of the digital aspect of chemistry that we get a better resolution for chemistry because we are actually forced to express things in a way that a computer can work with it in a meaningful way. That is semantics. It is philosophical, I have to say.
So is it true to say that the biggest challenge is figuring out how to structure the data?
Xiaochi Zhou: One of the biggest challenges is that when you describe something semantically, especially when you describe things on a large scale, it becomes very difficult for humans to access it. Usually, people need to construct formal representations, for example, a SPARQL query. Expertise in both domains is crucial; one must possess expertise in chemistry as well as an understanding of the knowledge graph’s underlying data structure to formulate accurate queries. Precision is important, even a misplaced comma could yield a very different result.
[Editorial: SPARQL, an acronym for SPARQL Protocol and RDF Query Language, is used to search and manipulate data structured in RDF (Resource Description Framework) format, which is a standardized model for presenting information on the Web. It allows querying of this data using a syntax similar to SQL (Structured Query Language) but customized for linked data. Linked data is a method for organizing and interconnecting information on the internet.]
This basically means it is nearly impossible for any user to construct a properly spelled query and only me and Alex can write something like this. So, we used natural language processing tactics to make the computer understand human language. It is a bit like an early version of ChatGPT. It uses language models to understand human questions and turn these into SPARQL queries, like “select” or “a molecule that has a boiling point below 50°”, things like that, and turn them into code.
I have been working on this for my whole Ph.D. and have made some very small progress in that field. So yes, putting chemistry data within a large graph solves a lot of problems, but it also introduces quite a number of new problems. Nevertheless, we are working on solutions.
Aleksandar Kondinski: Very often, you want to ask questions as general as possible while expecting precise and professional answers. This is particular challenging when dealing with something extensive like the whole field of chemistry like Xiaochi is doing. Ensuring accuracy and avoiding speculative responses or hallucinations, which is currently a concern with systems like ChatGPT, you can imagine would require a big effort.
Marie
Your system called Marie should be able to answer all chemistry questions. Can you describe what Marie is capable of at the moment?
Xiaochi Zhou: We have, so far, developed three versions. The first version uses quite a traditional language model. So, when a question is asked, it extracts the key components within the question. For example, for the question “What is the boiling point of benzene?” it identifies “benzene” as the target entity and searches the knowledge graph for what is called an IRI (Internationalized Resource Identifier) of the layer. This is like an IUPAC name for benzene, but it is a unique representation. There is an enlarged graph pointing to the entity benzene. From the question, “boiling point” is also identified and then converted to another IRI representing the boiling point property. By putting these two components, these two RIS, into a SPARQL template, it is quite easy to create a simple SPARQL query.
Then we realized that the knowledge graph might not contain everything you asked. So in the second version of Marine we added semantic agents. These are basically web services that can perform calculations or retrieve information from the knowledge graph. For example, we can ask a question like “What is the power conversion efficiency of a certain material?” and we have a semantic agent in the background that can specify the SMILES string (simplified molecular-input line-entry system; a specification in the form of a line notation to describe the structure of chemical species) of the material and calculate the corresponding conversion efficiency. Our language model is able to understand this question and launch a suitable agent to perform the calculation and return the data.
Marie 1 gives you the ability to look through the knowledge graph to retain the answer you want [4] and Marie 2 gives you the capability to calculate answers on the fly based on questions [5–7]. However, in the first two versions of Marie, we suffered from the speed problem. Typically, a SPARQL query on a large knowledge graph is slow; very, very slow.
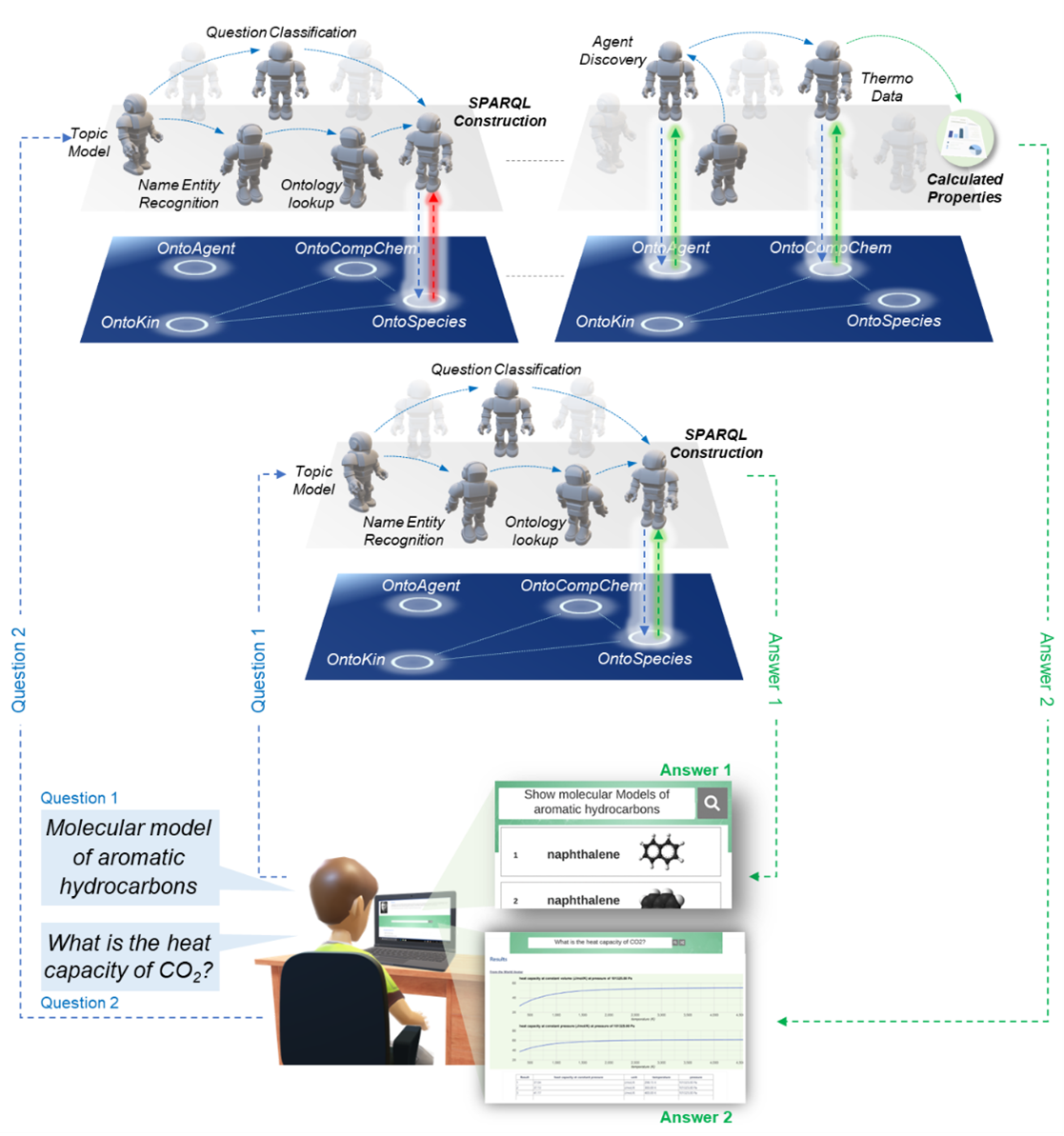
Figure 1. General implementations of Marie’s structure through an agent layer (grey) and a knowledge graph layer (blue).
In Marie 3, we have adopted a radical solution called knowledge graph embedding, which basically describes all relationships and properties within our chemical knowledge graph in a vector space. The idea is to parameterize all the knowledge into vectors and, by this, dramatically speed up information retrieval and also enable an operation called inference. This means you can derive unobserved facts based on observed facts.
For example, if I ask about the use of a particular chemical compound, that information might not have been explicitly described in the knowledge graph, but maybe we have already described the use of a very similar compound to that compound. The system could perform the inference, giving you an answer that is not even described in the knowledge graph.
That is Marie 3. It is a version that speeds up the query and also allows inference. The only disadvantage of Marie 3 is that it is quite expensive. It uses way more powerful language models. At that time, before ChatGPT, GPT3.5, and GPT4, BERT (Bidirectional Encoder Representations from Transformers; a family of language models introduced by researchers at Google in 2018) was the state of the art. We need to do embedding, a machine learning process. We need to build the entire knowledge graph, which consists of millions within billions of triples, and this process is computationally demanding. Despite the computational expense, the system works very well.
Now we are working on Marie 4. Everybody is talking about large language models—we have played with it and use them to directly come up with a SPARQL query based on questions.
Aleksandar Kondinski: This allows users to define more questions; but so many people have so many questions so it increases the demand.
Xiaochi Zhou: Yes, one of the biggest problems or challenges is that over years our users often go wild, asking about the meaning of life, the origin of the universe, …
… “Mirror, mirror on the wall, who’s the Fairest finest” —this is an actual question I found on the Marie log. I do not know who has asked this question nor why.
Can Marie answer all these questions? How does Marie differ from ChatGPT?
Xiaochi Zhou: One of the differences between a ChatGPT solution or a ChatGPT-like solution and our solution is that once our model does not have any idea about your question, it will tell you. “No, we don’t know about this. It is beyond our scope.” And even if it cannot provide an accurate answer, it would tell you something like “confidence for giving you an accurate answers is low”.
One of the biggest problems for large language models is hallucination, that it makes up a plausible answer based on your question even if it does not really understand your question or does not have an answer. However, our solution is to use language models to understand the question only, this model does not come up with an answer. It is the knowledge graph that provides you with the answer. This has two most important features:
First, the answer is variable and traceable. The system provides the answer to your question and you can trace it back within the knowledge graph. You can sort of see how the question and the answer are derived. It is not a black box model.
Second, because you can dynamically update your knowledge graph, you can keep your data fresh and up to date. A large language model may be trained every six months, so the answers might not be up to date.
Therefore, we are quite confident about our solution because it solves these two very important problems.
Why is the system called Marie?
Professor Kraft, our group leader, is a big fan of Marie Curie. She seems to be a very knowledgeable person and I think the copyright of her portrait has expired.
OntoMOPs
In comparison, how does OntoMOPs work?
Aleksandar Kondinski: Coming from polyoxometalate (POM) chemistry, researchers can guess, but they are not always 100 % sure what kind of compound will form, so they often try many different conditions. Systems like OntoMOPs help to reduce the uncertainty in inorganic synthesis because they give you an idea of which building blocks can be combined and which cannot.
In a paper we wrote that if you synthesized metal-organic polyhedra (MOPs) at random, you could derive 80,000 possibilities. However, how many of them can actually be produced? The real chemical space is not 80,000 possibilities. We found that you have 1,400 probable ways of getting structures. So if you go from 80,000 to 1,400, that is a huge reduction in waste [4].
How did work on OntoMOPs start?
Aleksandar Kondinski: When people started with knowledge models, they started with problems of retrosynthesis. You are looking for a way to build specific compounds in the lab.
OntoMOPs was our first step into solving chemical problems. I do not explicitly describe it as retrosynthetic, but you can also see it that way. The aim was not only to obtain a compound with a certain chemical composition and a certain type of functional group, but also that the compound has a certain shape or way of assembly.
The set of structures to develop this MOP discovery agent was not extremely high. We could look at something like a 150 different types of MOPs reported in the literature. From these, you can see that the overall chemical set is around 137 to 140 different chemical building units.
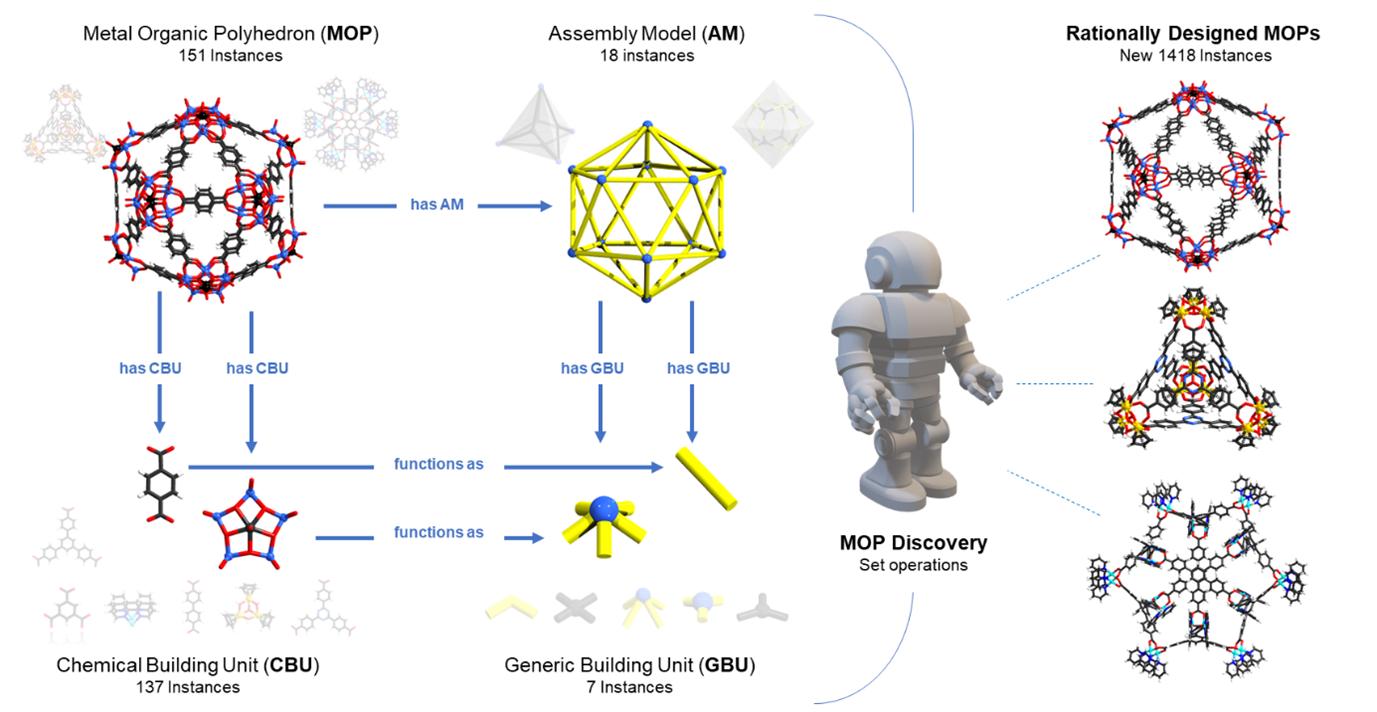
Figure 2. Assembly modelling as represented in OntoMOPs ontology (left) and the agent-based generation of new MOP formulations (right).
For us, at that point, it was important to verify that the new concept of assembly models and generic building units which I mentioned before is valid. Once this had been done, the work that we do has to do with polymers and other reticular materials such as POMs, MOPs, and metal-organic frameworks (MOFs). So we are now looking at different aspects where we can actually use more of the chemical way of saying, “Okay this kind of reaction can work, this kind of conversion is allowed, this kind of conversion is not allowed in this particular case,” and then basically make a pre-selection of building blocks.
Is that a system on its own or is it part of Marie?
Aleksandar Kondinski: Querying a knowledge graph requires the use of a query language such as SPARQL and an understanding of how the knowledge in that knowledge graph is structured, as Xiaochi described before. So the question arose as to how we can make this OntoMOPs system accessible to scientists through queries so that they can, for example, search for materials that have a certain property or a certain porosity. This is where Xiaochi’s work on Marie comes in.
Our work starts with small islands and these become more and more interconnected over time. But we have to start somewhere.
How does the OntoMOPs system effect competition, for example, in the area of reticular chemistry?
Aleksandar Kondinski: Many would like to patent and protect as many new compounds as possible. I have seen research groups that use some vague descriptions and basically try to patent the whole subject area. There are some patents like that.
I think the groups that are using AI are definitely going to try to come to innovation and protect it very quickly, and I think we have that competition right now. We are fueling it here, as well.
The difference between now and the past is that if you are the first in a field, you can patent or protect a lot more than you could before, so basically, every day matters a lot. When you read the patents and see how much is being protected, it is virtually more or less every molecule. I believe that some of the knowledge systems that are being developed can argue against this and make it necessary that you can maybe protect some molecules that you really made in your lab but not the whole field. But that is an open question. With AI we see some conflicts that have never appeared before. It is something like an uncharted area.
Credibility of Data
You said earlier that you can trace where the system gets the information from, so it is not a black box. Does this mean you can credit someone for having developed or synthesized something and correctly show the source of the data?
Aleksandar Kondinski: Ideally, you want to know who published, e.g., a synthesis, but also the one who verified whether a particular synthesis works. That is, there is a person who generated the data and there is someone who reviewed it and ensured that it was a correct source to input into the knowledge graph. These are two independent people. I am not sure what Xiaochi thinks about this. Currently we have a reference to the source, in most cases via the DOI.
Xiaochi Zhou: In knowledge graphs, we have qualifiers that annotate the data. For example, for the same data, for example, the boiling point of benzene, you might have different numbers from different sources. So in that case we will annotate different numbers and which number comes from which source. It is actually important to add this for the credibility of the resources.
Aleksandar Kondinski: There may be cases where even for humans it is very difficult to know which result is the correct one. People then tend to think, the system is not good enough to solve this problem. For example, you have two papers that are a little bit contradictory, and it might take years to resolve which is telling the truth. But with AI, people expect it should be able to solve something like that immediately and they have no understanding of why this can be difficult or impossible.
Future
Will systems like Marie change how we learn chemistry?
Aleksandar Kondinski: Probably in many ways. We should also not underestimate the possibility of playful learning. Sometimes people like to throw questions even to ChatGPT. They just want to see how it reacts. I think this may also be the possibility with Marie. You throw a question and you want to see what is known about it. It is not impossible that through this, people learn new things and also find gaps in knowledge. So playful learning plus finding a new domain to study.
How do you think the field will evolve?
Xiaochi Zhou: In my specific field, of course, everybody is talking about large language models, but I believe people have some practical illusions and expectations about using large language models to solve our problems. Because it is language-based and is not a fact-based model, there are some problems and challenges to overcome.
However, we do have found the potential of this particular tool in helping us to solve some problems. So I think chemistry data moves way closer to normal ordinary users, non-chemistry experts. For example, students in school, in a very near future—the technology would bring them closer to the data and knowledge will be never easier to access and understand.
First, I would say that the use of large models, not just large language models, will explode to solve problems in chemistry. Databases will increase because the cost of constructing large graphs and structured data is becoming lower and lower. Using language models, people are able to accurately convert abstract data such as reports, experimental results, papers, and other publications into structured data. Therefore, there will be a rapid growth of structured data in all fields, including chemistry and engineering.
What is your opinion Aleks?
Aleksandar Kondinski: I must say, that is a good vision. I think I will just add this: It will improve interoperability in chemistry between research groups and accelerate innovation by making it easier to understand what other communities are doing and to use some of their findings in your own research. I think this required a huge effort in the past.
Today people constantly see messages or some news on their phone. In the future, you may receive a message that a particular group has published this and how this might be relevant to your research. For example, there’s a new catalyst, but maybe you can reuse the principle in your framework, for example, for electron storage. In this case, I think we will definitely see a big innovation coming.
What about the costs of running such systems? You mentioned the easy access to knowledge, will everybody be able to access?
Xiaochi Zhou: The overhead costs for training sessions are quite high. The marginal cost is slightly lower and hosting such a service is not particularly expensive. I think the requirement for just hosting such a service is minimum. It is not significantly bigger than any other similar services.
It is quite expensive for us to build it.
What trends would you prefer not to see emerge?
Xiaochi Zhou: I prefer not to see, in the very near future, the use of large language models for interacting and answering questions by pouring chemistry data and publications into them and expecting to get reliable answers. While it might seem like an efficient approach, quoting Professor Kraft, all one has to do is “hammer it”. However, it consumes a lot of resources, money, and time. Although it appears magical to many, it just relies on the probability of words.
I also think proprietary models are harming the world. Our knowledge graph follows open data principles, we are more than willing to share it with other researchers and even businesses and to have community efforts to enhance and grow it. However, because of the high associated costs, people tend to not to share it. This might harm the entire ecosystem in this field. I am just trying to say, we are better people. [laughts]
Thank you very much for the very interesting insights.
References
[1] Aleksandar Kondinski, Jiaru Bai, Sebastian Mosbach, Jethro Akroyd, Markus Kraft, Knowledge Engineering in Chemistry: From Expert Systems to Agents of Creation, Acc. Chem. Res. 2022, 56(2), 128–139. https://doi.org/10.1021/acs.accounts.2c00617
[2] Vera Koester, Aleksandar Kondinski, Constructing Models for Teaching and Learning, ChemistryViews 2019. https://doi.org/10.1002/chemv.201900021
[3] Aleksandar Kondinski, Tatjana N. Parac-Vogt, Programmable Interlocking Disks: Bottom-Up Modular Assembly of Chemically Relevant Polyhedral and Reticular Structural Models, J. Chem. Educ. 2019, 96, 3, 601–605. https://doi.org/10.1021/acs.jchemed.8b00769
[4] Aleksandar Kondinski, Angiras Menon, Daniel Nurkowski, Feroz Farazi, Sebastian Mosbach, Jethro Akroyd, Markus Kraft, Automated Rational Design of Metal-Organic Polyhedra, J. Am. Chem. Soc. 2022, 144, 11713−11728. https://doi.org/10.1021/jacs.2c03402
[5] Xiaochi Zhou, Daniel Nurkowski, Sebastian Mosbach, Jethro Akroyd, Markus Kraft, Question answering system for chemistry, J. Chem. Inf. Model. 2021, 61(8), 3868–3880. https://doi.org/10.1021/acs.jcim.1c00275
[6] Xiaochi Zhou, Daniel Nurkowski, Angiras Menon, Jethro Akroyd, Sebastian Mosbach, Markus Kraft, Question answering system for chemistry—A semantic agent extension, Digital Chem. Eng. 2022, 3, 100032. https://doi.org/10.1016/j.dche.2022.100032
[7] Xiaochi Zhou, Shaocong Zhang, Mehal Agarwal, Jethro Akroyd, Sebastian Mosbach, Markus Kraft, Marie and BERT─ A Knowledge Graph Embedding Based Question Answering System for Chemistry, ACS Omega 2023, 8(36), 33039–33057. https://doi.org/10.1021/acsomega.3c05114
Aleksandar Kondinski studied chemistry at Jacobs University, Bremen, Germany. He obtained his Ph.D. in chemistry in 2016 at the same university under the supervision of Thomas Heine. From 2016 to 2017, he worked as a research associate at the Institute of Inorganic Chemistry at RWTH Aachen, Germany, in the research unit for molecular magnetism led by Paul Kögerler. From 2018 to 2020, he was a postdoc in the laboratory for Bioinorganic Chemistry, KU Leuven, Belgium, led by Tatjana N. Parac-Vogt.
Since 2020, he has served as a postdoctoral researcher in the group of Markus Kraft at the University of Cambridge, UK, and a Cambridge CARES Research Fellow at CREATE in Singapore.
Xiaochi Zhou studied electrical and electronic engineering at Nanyang Technological University (NTU), Singapore, and obtained his Ph.D. in 2023 in Chemical Engineering from Cambridge CARES at the University of Cambridge, UK, under the supervision of Professor Markus Kraft.
Also of Interest

Markus Kraft on the intersection of AI and chemistry, knowledge engineering and machine learning, their applications, challenges and successes, as well as his World Avatar Project

Discussing science communication, AI in chemistry, publication ethics, and the purpose of life with an AI

Alexei Lapkin, University of Cambridge, UK, on the state of the art and challenges of transforming chemistry into the digital realm
![]()


