
Vacuum/inert gas manifold systems, commonly called Schlenk lines, are ideal for handling compounds that react with O2, water, or CO2. Their design allows compounds to be handled and reactions to be performed under an inert atmosphere of argon or nitrogen. Specially adapted glassware with standardized Quick-Fit joints gives a highly modular and flexible system that enables a variety of experimental set-ups.
While Schlenk lines are conceptually simple, at first glance the mass of tubing and exotic glassware can be daunting for the first-time user. In part 1, we looked at basic Schlenk line design, some safety considerations, and how to start and stop a Schlenk line.
Here we look at the basis of all Schlenk line work, the evacuate-refill cycle, and how to dry and/or degas solvents. In future parts, we will present some basic experimental set ups for performing reactions with air-sensitive compounds and the isolation and analysis of your air-sensitive product(s).
The Vac and Back Cycle
The heart of any Schlenk line manipulation of air-sensitive compounds is the evacuate-refill cycle, also called the vacuum and backfill cycle, or just vac and back for short. This process removes the reactive atmosphere of the lab from the flask and replaces it with the inert one from the Schlenk line.
The application of vacuum, or evacuation of the flask reduces the amount of atmospheric gas in it to a fraction of the initial value. The flask is then filled with pure inert gas from the Schlenk line. The process is repeated at least three times as each evacuation further reduces the amount of the original atmosphere in the flask to Fn where F is the amount of atmospheric gases remaining after the first evacuation and n is the number of evacuation-refill cycles performed.
Preparing Vessels for Use on a Schlenk Line
All glassware should be clean and dry before use. As well as this, vessels need to have the reactive atmosphere of the lab replaced with the inert one from the Schlenk line. To do this, flasks are sealed with a greased, ground-glass stopper and evacuated, then filled with pure inert gas from the Schlenk line. The process consists of several steps and is repeated several times to ensure oxygen and moisture are sufficiently excluded.
Important! Make sure you have read the safety considerations in part 1 before commencing work.
To evacuate and refill a Schlenk tube
- Turn the Schlenk line on as described in part 1.
- Increase the gas flow so there are 2–3 bubbles per second in the bubbler. Be careful not to increase it too much or too quickly as you will blow oil out of the bubbler.
- Attach vessel to Schlenk line via tubing (Fig. 8, B–E). Use a wiggling or rocking motion to attach the tubing rather than a twisting motion (see part 1).
- Seal vessel with a greased, ground-glass stopper. Rubber septa should not be used as they do not hold a vacuum as well and can be sucked into the flask, exposing it to air. Rubber septa are also difficult to remove from inside a flask.
- Ensure tap H is open (Fig. 8).
- Open tap B to vacuum (Fig. 8).

- Wait 10–30 s depending on the size of the vessel. Larger vessels will take longer to evacuate.
- Close tap B.
- Open tap B slowly to gas.
- Watch the bubbler closely – the rate of the bubbles will slow and some oil will be sucked up the inner tube. Do not allow the oil to be sucked into the manifold.
- Close the tap as soon as you see the oil level rise up the inner tube and open it again more slowly.
- Repeat steps 9–11 until the vessel is completely filled with gas and the bubbler flow rate has returned to normal.
- Close tap B.
- Repeat steps 6–13 twice so that the vessel is evacuated and backfilled three times.
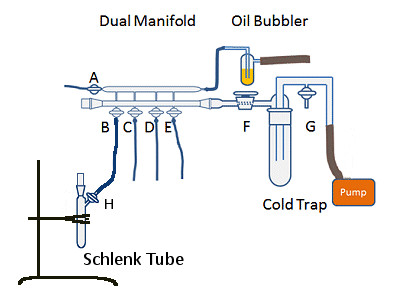
Figure 8. Simplified Schlenk line with flask ready to be evacuated and backfilled.
The real skill in this process is opening the vessel to gas slowly enough that you don’t suck oil into the manifold. If the oil is sucked into the manifold with sufficient force, it can contaminate reactions. More often, it leads to the person responsible having to clean the manifold – a fiddly and time-consuming task. Additionally, you will have sucked air back into the manifold, potentially contaminating your reaction.
Some bubblers are designed so that any oil sucked towards the line is collected and returned to the main body of the bubbler. These are a good idea, but can result in air being pulled into the line and contaminating anything open on the line, so care is still needed when opening a vessel to the gas line. If you have multiple vessels attached to the line while you backfill one vessel, it is a good idea to isolate them from the manifold by closing the appropriate taps. This will limit any contamination to the vessel you are working on, which can simply be placed back under vacuum and the process begun again.
As large vessels require more gas to fill them, they can be particularly problematic. If you do suck oil or air into your line, allow it to purge for a few minutes before resuming work.
Every time you connect something to the Schlenk line, the tubing and dead-space in the arm must be evacuated and backfilled 3–4 times before it is ready to use. This is especially important for flasks containing dry and degassed solvents, but also applies to reflux or distillation apparatus, solid addition flasks, and filter sticks.
Preparing Solvents
Drying Solvents
The lab atmosphere is not the only potential source of contamination by oxygen or water in your reaction. Solvents can contain sufficient CO2, O2, or water to ruin an air-sensitive compound if preventative steps are not taken. High purity solvent grades are available commercially; however, these can be substantially more expensive than the reagent grade equivalent. Depending on the quantity required, the cost of high purity solvents can be prohibitive and many labs in which air-sensitive chemistry is routinely performed will have permanent distillation stills for drying large quantities of commonly used solvents. These stills are run throughout the day so that freshly distilled solvent can be collected and used as needed.
If you need to dry a solvent yourself, addition of a desiccant is required (Tab. 2). The solvent should be of very low water content <0.1 % before being dried over a desiccant. This is achieved by azeotropic distillation, or by addition of a less active desiccant. The solvent should be allowed to stand over the less-active desiccant before being separated and distilled from a more active desiccant.
Table 2. Suitable drying agents for common solvents.
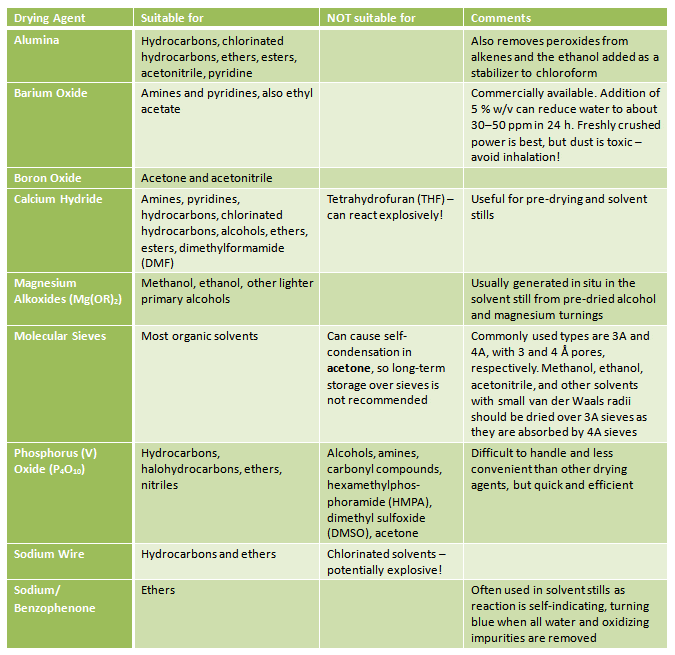
Detailed descriptions of solvent purification can be found, e.g., in Organic Solvents: Physical Properties and Methods of Purification by John A. Riddick, William B. Bunger, Theodore K. Sakano, 1986.
Due to the inherent dangers of having large quantities of boiling solvents in the lab, solvent stills are becoming less common. Instead, solvent purification systems that use a series of drying columns are finding their way into more and more labs. Use of these systems depends on the design, but generally, an evacuated Schlenk or Young’s flasks is attached directly to the column dispensing the desired solvent, the dead space in the connection must then be evacuated, finally, a tap is opened to allow the solvent to enter the flask.
Degassing Solvents
A dry solvent has only had one potential contaminant, water, removed. CO2, and O2 remain dissolved within the solvent if it has not been dried under an inert atmosphere or if you are using a high purity solvent straight from the bottle.
There are several methods you can use to degas or deoxygenate a solvent:
Purging
This is the simplest, but least effective method of degassing a solvent. It is suitable for roughly degassing large batches of solvent. It involves bubbling an inert gas through the solvent for 30 min–1 h, depending on the quantity of solvent to be degassed. The inert gas can be introduced to the solvent by a clean, long needle attached to the gas source. The needle can be attached via an adapter or via a 1-mL syringe that has had the plunger removed and the end cut off. This will fit neatly into the flexible rubber or plastic tubing of the Schlenk line. The needle should enter the vessel containing the solvent to be degassed through a rubber septum and the end of the needle should be positioned near the bottom of the solvent (Fig. 9). A pressure vent and exhaust, in the form of a short, disposable needle, should also be placed in the septum.
Care should be taken to ensure the flow rate of gas through the solvent is not too high as this can lead to increased evaporation of the solvent. A flow rate of 2–3 bubbles per second should be sufficient; however, this can be reduced for very low boiling solvents.
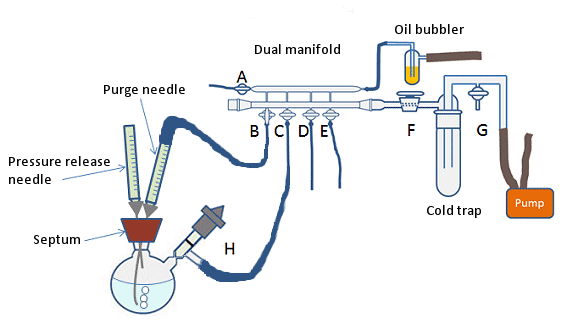
Figure 9. Degassing a solvent by purging with inert gas.
Atmosphere Exchange under Sonication
This technique is more effective than simple purging and can produce degassed solvents suitable for HPLC and some reactions very quickly. It involves sonication of the solvent while under light vacuum (i.e., house vacuum) and the refilling of the atmosphere with inert gas. The solvent is sonicated for up to a minute each time and the process is repeated 5–10 times.
Freeze-Pump-Thaw
This is the most effective method of degassing a solvent. It is suitable for smaller volumes of solvent, typically those that will fit in standard size reaction flasks. It involves freezing the solvent, evacuating the headspace and then allowing the solvent to thaw.
To degas a solvent by using the freeze-pump-thaw method:
- Fill Schlenk flask or thick-walled flask (suitable to withstand high vacuum) with the solvent to be degassed. Add a stirrer bar to flask if you wish.
- Attach flask to Schlenk line and ensure the tap H of the Schlenk flask (Fig. 10, step 1) is closed, i.e., the flask is sealed.
- Open line to vacuum (tap B), keeping the flask closed to vacuum.
- Place flask in a Dewar of liquid nitrogen until the solvent is completely frozen (Fig. 10, step 2).
- Open the tap H to the vacuum line and allow the headspace to be evacuated for 2–3 min (Fig. 10, step 3).
- Close the tap H on the flask, isolating flask from the vacuum (Fig. 10, step 4).
- Remove flask from liquid nitrogen and allow solvent to thaw (Fig. 10, step 5). A warm water bath on a stir plate can speed this process. The thawing of the solution under static vacuum allows any gas bubbles trapped in the solvent to escape into the headspace of the flask.
- Once the solvent is thawed, repeat steps 4–7. The freeze-pump-thaw cycle should be performed three times.
- After the third freeze-pump-thaw cycle, the flask can be refilled with the inert atmosphere of the Schlenk line. For this, open the line (tap B) and flask (tap H) to the inert gas slowly so as not to pull the external atmosphere back into the Schlenk line, as discussed above.
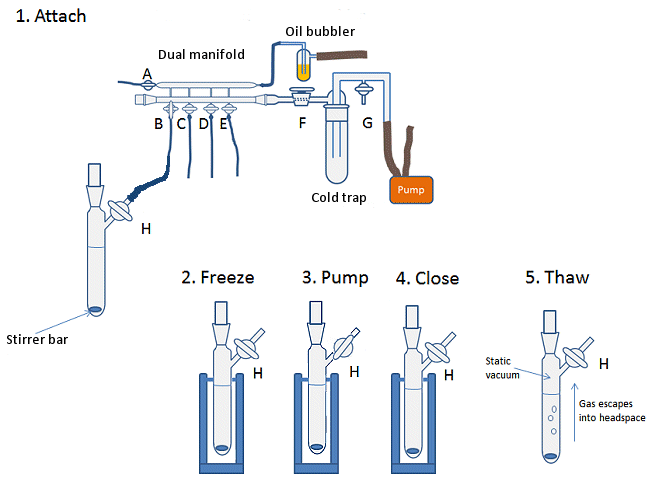
Figure 10. Degassing a solvent using the freeze-pump-thaw method.
Storing Dry and Degassed Solvents
Dry and degassed solvents must be handled and stored under an inert atmosphere for them to remain dry and degassed. Dry solvents can be stored over suitable drying agents, such as molecular sieves to prolong their life.
Solvents should be stored preferably in a Young’s flasks rather than a Schlenk flask. The Teflon tap of the Young’s flask provides a better seal against air contamination during storage. In contrast, the greased joint seal of a Schlenk flask can degrade over time, particularly in the presence of organic solvents and their vapors.
Long term storage is not recommended as impurities can be introduced through constant use. Small quantities of regularly prepared, fresh solvents are the only way to guarantee the solvent you use will not contain something that will ruin your air-sensitive reaction.
Finally, all members of the group using the dry solvents should be properly trained to make, store, open, use, and replace dry solvents. There’s nothing more irritating than tracing the cause of a failed – or successful! – reaction back to a solvent that someone has contaminated through carelessness. Especially when you have no idea what the contaminant is that is causing your reaction to fail or succeed!
Summary
The two interconnected lines of the Schlenk manifold provide a simple way to evacuate a flask and refill it with an inert atmosphere. This evacuate-refill cycle forms the basis of any air-sensitive technique performed on a Schlenk line. By repeatedly reducing the amount of external atmosphere to a fraction of its original value, water, O2, and CO2 can be sufficiently excluded to allow handing of air-sensitive compounds.
Likewise, any solvent being added to the reaction must have potential contaminants removed. Drying solvents over desiccants then degassing them is necessary before you can begin your reaction, which we will look at in part 3.
- Next month: Some basic experimental set ups for performing reactions with air-sensitive compounds and how to add solids and liquids.
- See all Tips and Tricks on air-sensitive chemistry
Further reading:
- Organic Solvents: Physical Properties and Methods of Purification,
John A. Riddick, William B. Bunger, Theodore K. Sakano,
John Wiley & Sons, New York, USA, 1986.
ISBN: 978-0-471-08467-9
Do you have any tip or tricks for handling air-sensitive compounds? Share them in the comments section …




The bubbler is simplified in the figure. As a practical guide, would it be better to show the details? The bubbler should contains either a check-valve or a buffer area to avoid oil being sucked back to the line, as illustrated here: http://prismresearchglass.com/catalog/product/gallery/id/6573/image/914/
Thank you for your comment. The link you provide nicely illustrates a bubbler in detail. Bubblers are also discussed in more depth in part 1 of this series: http://www.chemistryviews.org/details/education/3728881/Tips_and_Tricks_for_the_Lab_Air-Sensitive_Techniques_1.html