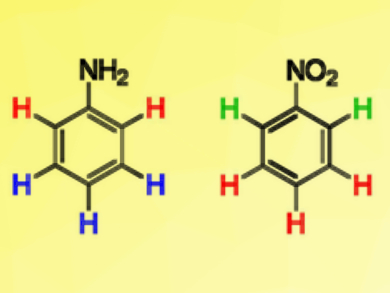
NMR spectroscopy is a powerful tool for structure determination. One of the basic properties giving insight into molecular structure is the signal position (chemical shift). Its rationalization is, thus, of great importance. π orbitals are commonly used to explain the high- or low-field shift of proton signals in benzene rings bearing an electron-donating or accepting group and the effect is associated with substituent resonance effects (+R/–R).
Using density functional theory (DFT) calculations, Marija Baranac-Stojanović, University of Belgrade, Serbia, has shown that the chemical shifts are not simply related to +R/−R effects. The proton chemical shifts of nitrobenzene (pictured right) are determined by the σ-electronic system, and by in-plane oxygen lone pairs in the case of ortho-H. In aniline (pictured left), the meta-H chemical shifts can be fully accounted for by the substituent’s +R effect, which also partly explains the para-H shift, while the higher-field position of ortho-H is fully determined by σ orbitals.
π orbitals are commonly invoked by chemists to explain various phenomena, such as the greater stability of conjugated versus nonconjugated systems, planarity, and chemical reactivity. This work, however, reveals an important role of the σ-electronic system.
- Can Variations of 1H NMR Chemical Shifts in Benzene Substituted with an Electron-Accepting (NO2)/Donating (NH2) Group be Explained in Terms of Resonance Effects of Substituents?,
Marija Baranac-Stojanović,
Chem. Asian J. 2018.
https://doi.org/10.1002/asia.201800137



