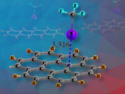
Being able to predict physical properties from a chemical structure is important for the development of self-assembled nanostructures, gels, and organic frameworks. Using fast, low-level computational techniques is an attractive way to make such predictions.
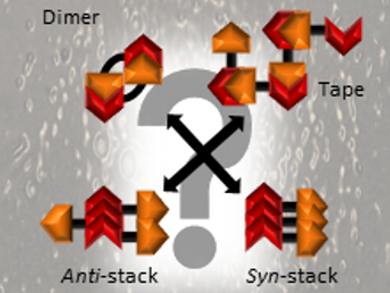
Jennifer Hiscock, University of Kent, UK, and colleagues have studied the hydrogen-bonded self-association of 39 structurally similar (thio)urea‐anion amphiphiles (compounds with both hydrophilic and lipophilic properties) within the solid, gas, and solution states. The team used experiments such as X-ray diffraction and NMR spectroscopy, as well as both high- and low-level computational modeling. Depending on the exact chemical structure and the conditions, the monomers can self-assemble into, e.g., tape-like structures, dimers, or different types of stacks (pictured).
The experimental and computational exploration of these compounds allowed the researchers to find structure-property relationships. Surface charge parameters from simple low-level computational modeling techniques were found to correlate well with experimentally derived solution-state dimerization constants. A correlation was also identified between experimentally derived CMC (critical micelle concentration) values and calculated surface charge and LogP (partition coefficient) parameters. Together, these correlations demonstrate the potential for low-level modeling to predict the physical properties of self-associated systems.
- Towards the Prediction of Global Solution State Properties for Hydrogen Bonded, Self-Associating Amphiphiles,
Lisa J. White, Stilyana N. Tyuleva, Ben Wilson, Helena J. Shepherd, Kendrick K. L. Ng, Simon J. Holder, Ewan R. Clark, Jennifer R. Hiscock,
Chem. Eur. J. 2018.
https://doi.org/10.1002/chem.201801280