The chemistry of ferrocene, a sandwich complex composed of two cyclopentadienyl ligands and a central iron atom, has been explored extensively. The isoelectronic cobaltocenium cation is not as well-studied. In contrast to the electron-rich ferrocene, cobaltocenium generally does not undergo electrophilic aromatic substitutions, but rather nucleophilic addition reactions at a cyclopentadienyl ligand.
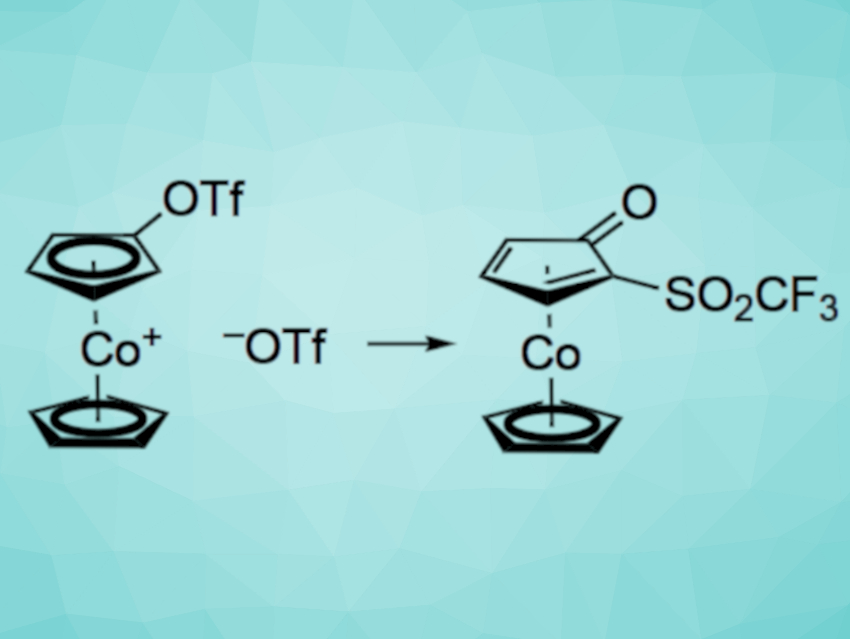
Holger Butenschön and Geanne M. R. Boston, Leibniz Universität Hannover, Germany, have developed the first anionic thia-Fries rearrangement at a cobaltocenium cation. This type of reaction involves the conversion of an aromatic triflate to a rearranged product. In this specific case, an unprecedented ortho-deprotonation of a (trifluoromethylsulfonyloxy)cobaltocenium cation using the non-nucleophilic base lithium diisopropylamide (LDA) was followed by a 1,2-migration of the trifluoromethylsulfonyl group (pictured, Tf = SO2CF3). As a result, a 2-(trifluoromethylsulfonyl)cyclopentadienone ligand stabilized in the form of a cyclopentadienylcobalt complex was formed.
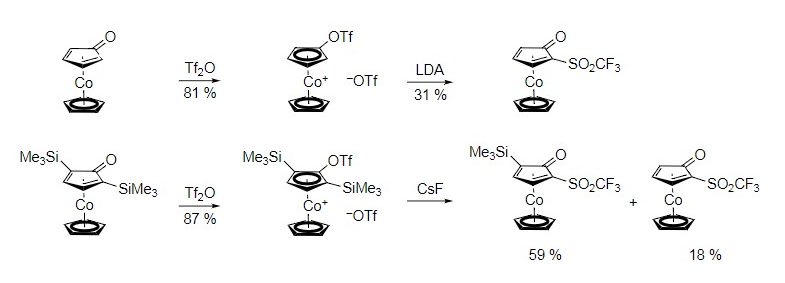
The desilylation of the corresponding silyl derivatives with cesium fluoride (pictured) also leads to rearrangement products. This reaction opens a new path to substituted cyclopentadienones, which can be freed from their cobalt complexes via oxidation and could be promising building blocks for organic synthesis.
- The First Anionic Thia‐Fries Rearrangement at the Cobaltocenium Cation,
Geanne M. R. Boston, Holger Butenschön,
Eur. J. Inorg. Chem. 2022.
https://doi.org/10.1002/ejic.202200143
![]()


