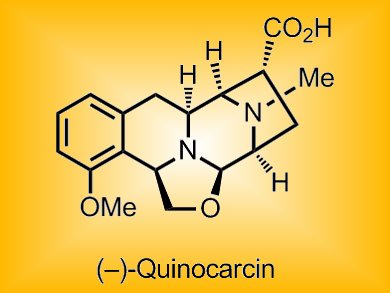
Tetrahydroisoquinoline alkaloids are a structural class of natural products, many of which show promising anticancer activity, with a few in advanced clinical trials. One member of this class, (–)-quinocarcin, shows remarkable antiproliferative activity against lymphocytic leukemia; its more stable citrate salt (KW2152) and the aminonitrile-substituted analogue DX-52-1 show inhibitory activity towards non-small cell lung cancer and adenocarcinoma. Thus, this class of alkaloids is of considerable interest because of their potential use as anticancer agents. Although synthetic methods have been reported, no convergent route providing general access to the entire compound class has been found.
Nobutaka Fujii, Hiroaki Ohno, and co-workers, Kyoto University, Japan, have achieved an asymmetric total synthesis of quinocarcin through a convergent strategy based on Sonogashira coupling and gold(I)-catalyzed hydroamination. The troublesome regioselectivity (5-exo–dig versus 6-endo–dig, pictured) of the intramolecular hydroamination reaction was resolved by substrate-controlled selectivity inversion and use of a chlorine-mediated benzofuran cleavage.
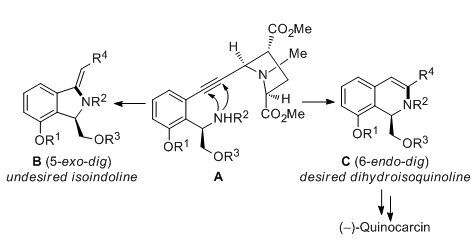
These findings could lead to the development of a general and divergent synthetic strategy for related tetrahydroisoquinoline alkaloids.
- Convergent Synthesis of (–)-Quinocarcin Based on the Combination of Sonogashira Coupling and Gold(I)-Catalyzed 6-endo–dig Hydroamination,
Hiroaki Chiba, Yuki Sakai, Ayako Ohara, Shinya Oishi, Nobutaka Fujii, Hiroaki Ohno,
Chem. Eur. J. 2013.
DOI: 10.1002/chem.201300687