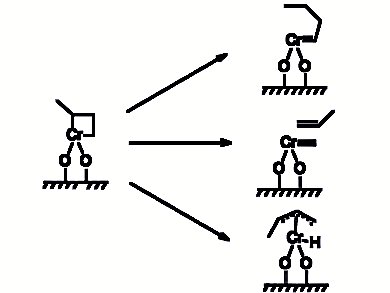
Ethylene Polymerization Over the Phillips CrOx/SiO2 Catalyst
In the paper “Active Site Transformation During the Introduction Period of Ethylene Polymerization over the Phillips CrOx/SiO2 Catalyst”, Liu et al. [1] proposed the stable geometries of a CrII cluster model as precusor for the polymerzation process. It involves a highly coordinatively unsaturated CrIIOx, surface species and the lone-paired electrons of the oxygen atom of adsorbed formaldehyde.
It is a contribution that is worth reading, as it enriches the previously proposed mechanisms for the initiation step of ethylene polymerization [2]. Density functional theory (DFT) calculations were adopted to achieve a basic understanding of the effect of formaldehyde coordination and desorption on the active site transformation during the induction period at a molecular and atomic level (Fig. 1) [1].
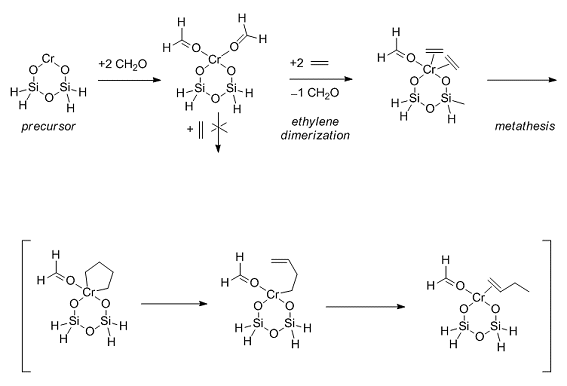
Figure 1. Reaction pathway for ethylene polymerization as proposed by Liu et al. [1].
The Structure of Catalytic Active Sites
Exploratory work focusing on the catalytic active site has its origins in the early 1960s and has continued up until now in an untiring competition in academia. The discussion of active sites in the ethylene polymerization over CrOx/SiO2 was narrowed down by the “advance” in understanding of mechanistic details in the Ziegler–Natta system; the formation of an active site supplying a metal–carbon or metal–hydride bond is followed by chain propagation, based on the proposed Cossee mechanism [3]. But chain propagation could not really answer the question about the source of hydrogen needed.
During the initial period of ethylene polymerization in the absence of an activator, CrVI is reduced to a lower valance state, therefore, hydrogen, as well as other reduction products are used to fulfil the formal stoichiometry. It was also speculated that Si–OH groups may be acting as a hydrogen source.
As only a minor portion of the CrVI supported on silica is involved in the polymerization process, different questions have come up: Which is (are) the relevant valance state(s)? What kind of intermediates can be analyzed as a result of the induction period?
Based on these high demands, analytical techniques should differentiate between catalytically active sites and the remaining inactive sites. But spectroscopic techniques, however sophisticated, are sometimes inadequate for this purpose, and some spectra are still misleading [1].
Proposed Reaction Pathway
Krauss and Stach found that CO quantitatively reduces surface chromate at 350 °C to chromium (II), which is believed to be the catalytic active valence state of the Phillips catalyst [4]. This finding stimulated the DFT investigation by Liu et al. [2].
As a result of preparation of chromium-supported silica, discussion about the activated contact was focused on mono, dichromate, and polychromate structures. Preparation was performed by wet impregnation, starting from chromium hexavalent oxide to form mono and/or dichromate esters, but was limited by the amount of available surface silanol groups of the support. If the ratio is well balanced, no Cr2O3 should be formed after the material is activated at 800 °C. Krauss et al. [5, 6] showed that activation and reduction temperatures are important factors to control reactivity [7, 8].
According to the findings, the CrII species is highly coordinatively unsaturated for molecules that can be adsorbed, but depends on calcination/reduction temperatures. Different classes of CrII species are proposed to exist with at least two, one, or no free coordination sites based on CO as probe molecule. These are marked as CrA, CrB, and CrC, respectively. These are distinguished by the degree of coordination to the silica surface and caused by hydroxy and/or silxane groups on the surface.
In this context, one or two additional oxygen atoms may be coordinated to CrB and CrC, respectively, created by surface siloxane (Si–O–Si) or silanol (Si–OH) groups. These were analyzed by temperature-programmed desorption–temperature-programmed oxidation (TPD–TPO), IR, and UV/Vis spectroscopy [7–10]. CrA was identified as the most catalytic active species that is active for the ethylene polymerization, whereas CrC was found to be completely inactive.
Thermal activation of surface hydroxy groups increases the amount of siloxane groups, whereas at the same time the catalytic active sites become more active for ethylene polymerization, which can, therefore, be controlled and used by industry.
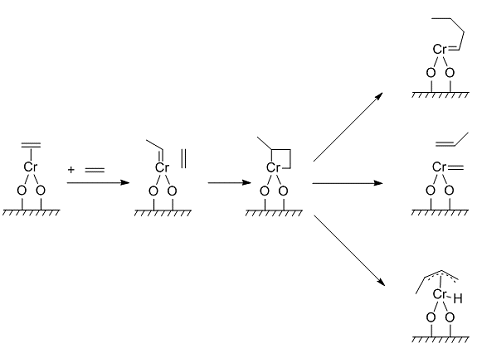
Figure 2. Reaction pathways via C1 species (carbene complex formation and chain propagation) as proposed by H. L. Krauss, E. Hums [23, 11].
The formation of an allyl species, proposed by Krauss [11], should exclusively apply to CrA and polymerization might either start from a H or allyl coordinated species (see Figure 2); it is assumed that a carbene intermediate is involved [12–14]. A chain-transfer reaction terminates the chain and allows a new one using the same active site. Finally a transfer then occurs through β-hydride elimination [15], resulting in α-olefins with even and uneven numbers of carbons, which are identified by capillary gas chromatography. At the same time, this reaction is accompanied by the formation of small amounts of lower alkanes, such as CH4, C2H6, and in low concentrations C3H8 and n-C4H10. The source of the hydrogen was proven by using deuterated silica, which resulted in the production of CH2D2 and C2H4D2 [16, 17].
A further aspect that should not be neglected is the topology of silanol groups. Differential thermal gravimetric/thermogravimetric analysis (DTG/TG) and IR spectroscopy showed maxima for three topologically privileged arrangements of silanol groups that differ in their ratio for specified gels [18]. In contrast, calculations presented by Liu et al. [1] are restricted to a well outbalanced geometry of an ideal plane of a six-membered CrII siloxane ring. To modify the resulting binding energies and geometry parameters by the number of adsorbed formaldehyde molecules. It demonstrates the elucidation of the rather thermodynamically stable cluster models, which were the starting point in the study of steric hindrance based on the intermediates involved.
If Demmelmaier et al. [19] are right in proposing that chromate formed on silica pretreated at 200 °C is unable to initiate ethylene polymerization, whereas chromate formed on silica pretreated at 800 °C shows ethylene polymerization activity, the question must come up, can these differences be noticed by DFT calculations?
Limits of DFT Calculations
In our studies, we experienced a number of data sets when NO decomposition on Cu sites was performed by DFT calculations. Although we could describe the reaction path via intermediates within proposed cluster models we did not get corresponding know-how to advance in experimental improvements [20, 21]. This finally showed us the crux of bridging the gap of reaction and molecular models, as well as the sensation of pressure that reaction models and molecular models must grow together.
It is of great interest that Espelid and Børve studied several cluster models of CrIIOx, surface species covering a range of O–Cr–O angles by using DFT [22]. They draw the conclusion that a cationic Cr–C species is formed by protonation of Cr=C (carbene). This was shown by a lower reaction energy barrier than the metallacyclic or Green–Rooney carbene propagation [22], thus suggesting the transformation of Cr=C (carbene) into Cr–C sites to confirm the proposal of Cossee [3]. When treating the contact in a deuterium stream, CH3D is formed [23]. But in contrast to Espelid and Børve’s findings [22], no activity was observed before. Therefore we have to conclude that Cr–C sites do not show us an active behavior for the polymerization process.
It should also be mentioned that the proposed equilibrium between the metalla four-membered ring and the π-allyl metal complex is applicable if cyclic monoolefins with more than six carbon atoms in the ring are used. A ring contraction down to C6 isomers of simple- and double methylated cycloolefins can be found as a result in separate runs of experiments to study the C1 transfer. The hydrogenation of rings and their derivatives shows that hydrogen is a floating part of the catalytic system. However, the transfer is confirmed even in a reaction with methyl cycloolefins [23].
As the reaction on CrVIOx–SiO2 with ethylene resulted in the formation of some non-olefinic products such as CO, CO2, H2O, H2, CH4, and C2H6, as well as formaldehyde, the initial period of the ethylene polymerization does also show Fischer–Tropsch behavior. It is believed that both reactions can be formulated over an instable CrA formaldehyde complex [24]. From this point of view theoretical modeling based on the interaction of formaldehyde published by Liu et al. [1] should be also open to these results to surmount a self-contained picture of the isolated CrII siloxane ring species.
Because computing capacity has meanwhile greatly improved, all experimental data should finally find a corresponding molecular model. Hierarchical ranking of the complex experimental data should draw molecular modeling much closer to the facts as discussed above.
If we agree exclusively with the cluster model CrIIOx, surface, which is the result of the DFT calculations, it would be nearly impossible to show experimental evidence of an isolated CrII siloxane ring that is coordinated to one formaldehyde molecule with regards to the assumed products, such as oligomeric and/or polymer products. Therefore, it is a must to include the silica matrix to modify the presented results of the DFT calculations.
References
[1] L. Zhong, Z. Liu, R. Cheng, S. Tang, P. Qiu, X. He, M. Terano, B. Liu, Active Site Transformation During the Induction Period of Ethylene Polymerization over the Phillips CrOx/SiO2 Catalyst, ChemCatChem 2012, 4, 872–881. DOI: 10.1002/cctc.201100278
[2] M. P. McDaniel, A Review of the Phillips Supported Chromium Catalyst and Its Commercial Use for Ethylene Polymerization in: Advances in Catalysis (Eds. Bruce C. Gates, Helmut Knözinger), Elsevier, Amsterdam, 2010, 53, 123–606. DOI: 10.1016/S0360-0564(10)53003-7
[3] P. Cossee, J. Catal. 1964, 3, 80–88. DOI: 10.1016/0021-9517(64)90095-8
[4] H. L. Krauss, H. Stach, Z. Anorg. Allg. Chem. 1969, 366, 280–290. DOI: 10.1002/zaac.19693660505
[5] H. L. Krauss, B. Rebenstorf, U. Westphal, D. Schneeweiß, in: Proc. Int. Symp. on Prep. of Catalysts (Eds. B. Delon, P. A. Jacobs, G. Poncelet), Elsevier, Amsterdam, 1976, 489–495.
[6] H.L. Krauss, R. Höpfl, Proc. Eur. Sym. Therm. Anal. (PESAG) 1981, 2, 175.
[7] G. Ghiotti, E. Garrone, A. Zecchina, J. Mol. Catal. 1988, 46, 61–77. DOI: 10.1016/0304-5102(88)85083-1
[8] H. L. Krauss, B. Rebenstorf, U. Westphal, Z. Anorg. Allg. Chem. 1975, 414, 97–108. DOI: 10.1002/zaac.19754140202
[9] M. Nishimura, J. M. Thomas, Catal. Lett. 1993, 21, 149–155. DOI: 10.1007/BF00767380
[10] C. S. Kim, S. I. Woo, J. Mol. Catal. 1992, 73, 249–263. DOI: 10.1016/0304-5102(92)80077-T
[11] H. L. Krauss, in: Symposium on the Mechanisms of Hydrocarbon Reactions (Eds. F. Marta, D. Kallo), Elsevier, Amsterdam, 1975, 227–233.
[12] H. L. Krauss, E. Hums, Z. Naturforschung 1979, 34b, 1628–1636.
[13] K. J. Ivin, J. J. Rooney, C. D. Stewart, M. L. H. Green, R. J. Mahtab, J. Chem. Soc. Chem. Commun. 1978, 323, 604–606. DOI: 10.1039/C39780000604
[14] G. Ghiotti, E. Garrone, S. Coluccia, C. Morterra, A. Zecchina, J. Chem. Soc. Chem. Commun., 1979, 801, 1032–1033. DOI: 10.1039/C39790001032
[15] M. P. McDaniel, M. M. J. Johnson, J. Catal. 1986, 101, 446–457. DOI: 10.1016/0021-9517(86)90272-1
[16] H. L. Krauss, E. Hums, Z. Naturforschung 1980, 35b, 848–854; 1983, 38b, 1412–1418.
[17] H. L. Krauss, E. Hums, Z. Naturforschung 1983, 38b, 1412–1418.
[18] D. Naumann, Thesis: Eigenschaften und Struktur von CrVI-, CrII– und CrIII-Oberflächenverbindungen unter dem Aspekt der Topologie des amorphen Trägers “Silicagel”, Freie Universität Berlin, Germany, 1979.
[19] C. A. Demmelmaier, R. E. White, J. A. van Bokhoven, S. L. Scott, J. Catal. 2009, 262, 44–56. DOI: 10.1016/j.jcat.2008.11.024
[20] T. Clark, E. Hums, J. Schamberger, Abstracts of the Second International Memorial G. K. Boreskov Conference, Novosibirsk, Russia, 1997, Part II, 383.
[21] E. Hums, Catal. Today 1998, 42, 25–35. DOI: 10.1016/S0920-5861(98)00073-X
[22] Ø. Espelid, K. J. Børve, J. Catal. 2000, 195, 125–139. DOI: 10.1006/jcat.2000.2986
[23] E. Hums, Ind. Eng. Chem. Prod. Res. Dev. 1985, 24, 517–520. DOI: 10.1021/i300020a006
[24] E. Hums, 20. Hauptversammlung der GDCh, Heidelberg, Germany, September 16–19 1985.
Dr. J. E. Hums
Consulting Environmental Catalysis, Erlangen, Germany



