New Challenges
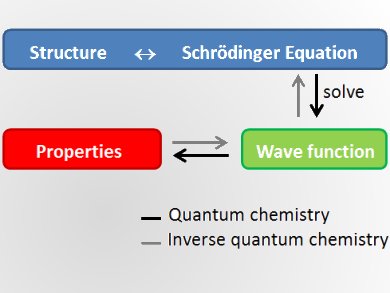
Quantum chemistry uses the Schrödinger equation to predict the properties of given molecular structures. It started in 1927 with the first true calculation of the hydrogen molecule. In 1970, the first commercial quantum chemistry codes were used to study molecules, their properties, and how they act together in chemical reactions. Nowadays quantum chemistry is used in all branches of chemistry as well as in physics and biology. Quantum chemical studies begin from a structure, make approximations, and predict the properties of this molecule.
According to Markus Reiher, ETH Zurich, Switzerland, a new challenge of quantum chemistry is to turn this concept around and calculate a molecular structure with defined properties. He thinks it is possible that in a few years scientists will design materials by looking at the properties they want and then predict which structures feature these properties. If feasible, this would challenge extensive screening and time- and cost-intensive trial-and-error synthesis processes, which are currently the state of the art in the field. This would completely change compound design.
Inverse Quantum Chemistry
Inverse quantum chemistry starts from the properties to calculate a suitable molecule. Formally, this means to invert the Schrödinger equation. Unfortunately, this is mathematically ill-defined, because it is an ambiguous assignment with many potential structures for one property. Still, Reiher believes that for specific well-defined problems the inversion can be achieved, particularly if chemical heuristics can be computationally exploited.
Traditional sampling methods have to look at large subsets of the chemical space, which is the space of all molecules. Inverse quantum chemistry is only possible if the search is limited to small areas of the chemical space.
In a recent publication in the International Journal of Quantum Chemistry, Markus Reiher discussed several inverse quantum chemistry applications proposed so far [1]. The basic idea of inverse quantum chemistry was presented as early as the seventies by G.G. Hall. Today, advances in computer technology have made it possible to solve increasingly complicated problems. This also makes real-time calculations and thus interactivity possible [2]. But it is only with new concepts and simplifications that the vision of inverse quantum chemistry will become a reality within the near future.
From the 1920s to the late 1960s, it was barely possible to handle the complicated mathematical equations of quantum mechanics for molecules. Today more or less every chemist can solve the Schrödinger equation, and quantum chemistry has emerged as its own branch of chemistry. We owe this to, for example, Hartree and Fock who in 1928/1930 introduced the central approximation that instead of treating the motion of each electron separately, treates them as if each particle is subjected to the mean field created by all other particles; and to Hohenberg and Kohn who in 1964 laid the basis for the density functional theory (DFT). DFT is currently the most commonly used quantum chemistry concept that is capable of treating relatively large systems at acceptable computational costs [3, 4].
In 1951, Roothaan and Hall formulated independently of each other the basis set of approximation, which allows the efficient calculation of molecular orbitals and energies – such as in the Gaussian program. For the first successful implementation and application Pople was awarded the Nobel Prize in Chemistry jointly with Kohn in 1998.
Proof of Principle
Markus Reiher and his team recently started a pilot study to try to rebuild the Schrock catalyst for nitrogen fixation by using inverse quantum chemistry [5]. On the basis of DFT, they created a new concept for rational compound design, namely, gradient-driven molecule construction (GdMC).
Looking for an N2 binding complex, they started with a predefined central fragment such as one Mo that should bind N2. For a stable structure the energy gradient should be zero, but this is not the case for the small starting fragment that instead exhibits forces on all atoms that drive the metal atom and the dinitrogen ligand apart.
Then they surrounded this fragment by a potential called the jacket potential, which minimizes the gradient. In a second step, the jacket potential is replaced by a chemically viable chelate-ligand structure that stabilizes the fragment. For a stable complex geometry the gradients on all of its atoms are required to go to zero.
Although it is not straightforward to optimize the jacket potential, it is possible to construct the chelate ligand by relying on the concept of functional groups. Other optimization strategies were investigated in detail and show a broad potential for future explorations. By this the team is able to present very first steps towards the design of a new transition-metal catalyst for the homogeneous fixation of dinitrogen.
Outlook
Reiher believes that such novel approaches will be needed in quantum chemistry to surpass its past achievements. Moreover, it is needed to promote this research field beyond a set of techniques available in computer programs like Gaussian that can be used by non experts.
“Chemistry is involved with solving many of the most pressing problems of humanity. New molecules can take us far. Markus has made an important step to make inverse design more practical. I hope one day methods like his are commonplace and available for everyone to use in popular chemistry software packages.” Alan Aspuru-Guzik, Harvard University, Cambridge, MA, USA, told ChemViews Magazine.
According to Markus Reiher, “It is only a question of time until the first ideas that have been explored in inverse quantum chemistry so far will produce a truly novel result that will demonstrate their true capabilities. Once this has been achieved, many others will start working on this”.
References
[1]
Inverse Quantum Chemistry: Concepts and Strategies for Rational Compound Design,
T. Weymuth, M. Reiher,
Int. J. Quantum Chem. 2014, 114, 826–837.
DOI: 10.1002/qua.24687
[2]
M. P. Haag, A. C. Vaucher, M. Bosson, S. Redon, M. Reiher,
ChemPhysChem 2014.
DOI: 10.1002/cphc.201402342 (in press)
[3]
Frank Jensen,
Introduction to Computational Chemistry,
Wiley, 2006.
ISBN: 978-0-470-01187-4
[4]
Cristopher J. Cramer,
Essentials of Computational Chemistry: Theories and Models,
Wiley, 2004.
ISBN: 978-0-470-09182-1
[5]
![]() Gradient-Driven Molecule Construction: An Inverse Approach Applied to the Design of Small-Molecule Fixating Catalysts,
Gradient-Driven Molecule Construction: An Inverse Approach Applied to the Design of Small-Molecule Fixating Catalysts,
T. Weymuth, M. Reiher,
Int. J. Quantum. Chem. 2014, 114, 838–850.
DOI: 10.1002/qua.24686