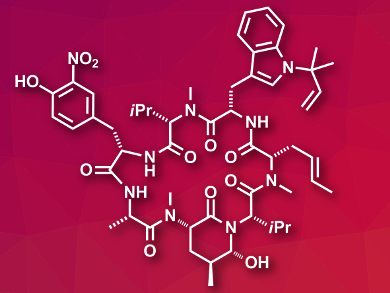
Tuberculosis (TB) is a global health problem. Current Anti-TB drugs often have severe side effects, and additionally, multi-drug-resistant TB is a rising problem. New treatment options would, thus, be useful. Ilamycin E1 (pictured) is a cyclopeptide isolated from marine bacteria with a very potent anti-tuberculosis activity. This natural product could serve as a lead compound for drug development.
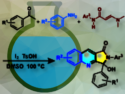
Yian Guo, Tao Ye, and colleagues, Peking University, Shenzhen, China, have performed the first total synthesis of ilamycin E1 and the related compound ilamycin F. The team used a convergent strategy, in which two fragments of the final compound were connected by an amidation and a macrolactonization to give the cyclopeptide structure. One fragment, a tripeptide, was synthesized starting from tryptophan in five steps, and the other, a tetrapeptide, was synthesized from glutamic acid in thirteen steps.
After the final lactonization, the cyclic product could be converted to ilamycin E1 in two steps, which form the target compound’s piperidone unit. The same cyclic product can also be converted to ilamycin F by a different two-step process using a Pinnick oxidation. Interestingly, this last reaction can also be reversed, i.e., ilamycin F can be converted into a precursor for the synthesis of ilamycin E1.
Since ilamycin F is more abundant in nature, but ilamycin E1 is much more potent against TB, this is a particularly useful discovery. Overall, these developments can provide sufficient amounts of ilamycin E1 for biological studies.
- Total Synthesis of Anti-tuberculosis Natural Products Ilamycins E1 and F,
Yingying Cheng, Shoubin Tang, Yian Guo, Tao Ye,
Org. Lett. 2018.
https://doi.org/10.1021/acs.orglett.8b02643