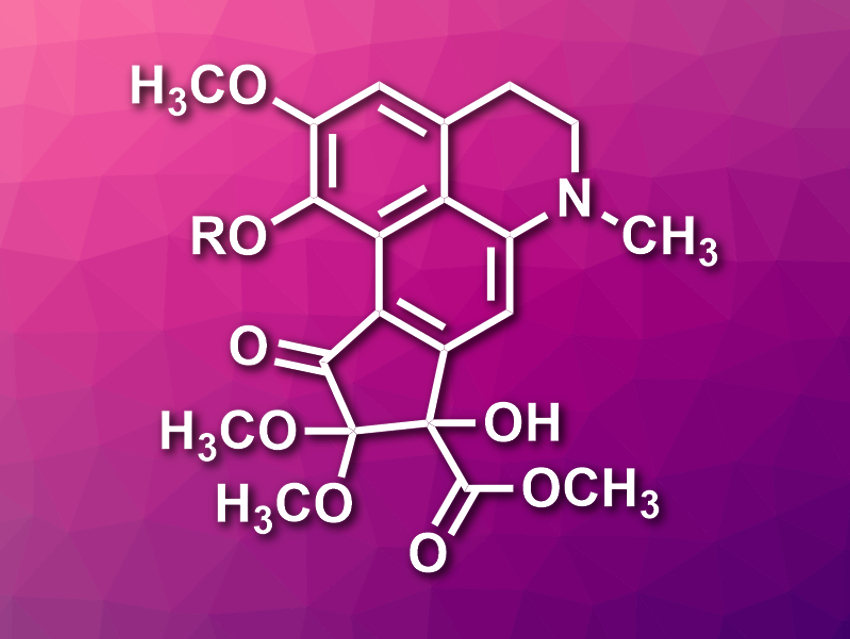
Dactylicapnosines A and B are two natural products isolated from Dactylicapnos scandens, a herb used in traditional Chinese medicine. The tetracyclic compounds can be classified as 9,10-seco-7-dehydroaporphines and only differ in one residue (pictured, R = CH3 for dactylicapnosine A, R = H for dactylicapnosine B). The compounds have interesting biological activities; dactylicapnosine A, in particular, shows significant anti-inflammatory activity. However, the total synthesis of dactylicapnosine A had only been achieved in 5 % overall yield so far, and no total synthesis of dactylicapnosine B had been reported.
Xiao-Dong Luo, Yunnan University, China, and Kunming Institute of Botany, Chinese Academy of Sciences, Hongbin Zhang, Yunnan University, and colleagues have developed an improved synthesis of dactylicapnosine A and achieved the first total synthesis of dactylicapnosine B. The team started from 2,3,4-trimethoxybenzaldehyde, which was converted to a phenol via a Baeyer–Villiger oxidation, followed by an allyl ether formation and a Claisen rearrangement to give an allylated derivative. Ozonolysis was used to convert this allyl group to an aldehyde, which was reacted with an amine to give a tetrahydroisoquinoline intermediate.
A palladium-catalyzed coupling reaction was used to close the fourth ring in the compound, and the phenol unit was converted to a para-quinone. The desired five-membered ring was formed via a cobalt-mediated ring-contraction reaction of this quinone. With the overall skeleton of the desired compounds in hand, further oxidation, deprotection, and methylation steps gave dactylicapnosine A in an overall yield of 12 %. Dactylicapnosine A was then converted to dactylicapnosine B via a reaction with boron trichloride, leading to an unstable diketone intermediate, followed by a methoxylation using 2,2-dimethoxypropane.
- Total Synthesis of Dactylicapnosines A and B,
Yinjiao Zhao, Yuda Li, Bei Wang, Jingfeng Zhao, Liang Li, Xiao-Dong Luo, Hongbin Zhang,
J. Org. Chem. 2020.
https://doi.org/10.1021/acs.joc.0c01900
![]()