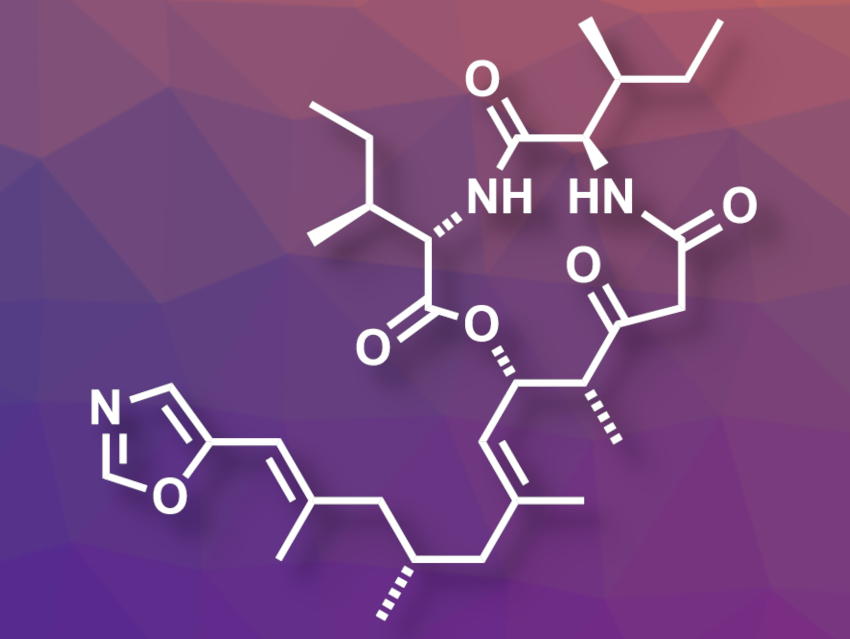
(+)-Taumycin A (pictured) is a biologically active macrolide isolated from a marine sponge. While the stereochemistry of the twelve-membered ring is known, the absolute configuration at the methyl-substituted C(9) (in the sidechain at the bottom of the picture) had not been assigned so far.
William A. Maio, New Mexico State University, Las Cruces, USA, and colleagues have performed an asymmetric total synthesis of (+)-taumycin A. The team’s approach targeted both C(9) diastereomers and allowed them to assign the stereochemistry at this carbon atom. They first prepared quasi-enantiomeric precursors of the sidechain with opposing configurations at C(9) and orthogonal protecting groups at the alcohol ends. These protecting groups were then removed separately to obtain the corresponding free alcohols, each serving as a precursor for one of the targeted diastereomers.
Each free alcohol was oxidized and reacted with (R)-(−)-4-benzyl-3-propionyl-2-oxazolidinone. Then the team installed β-ketoester groups and dipeptide units, followed by a cyclization to obtain the desired diastereomeric products, C(9)-R-(+)-taumycin A and C(9)-S-taumycin A. The team found that the spectral properties of C(9)-S-(+)-taumycin A were closest to those of the natural product.
- Asymmetric Total Synthesis and Revision of Absolute Stereochemistry for (+)-Taumycin A: An Approach that Exploits Orthogonally Protected Quasienantiomers,
Uttar K. Shrestha, Alexandra E. Golliher, Tara D. Newar, F. Omar Holguin, William A. Maio,
J. Org. Chem. 2021.
https://doi.org/10.1021/acs.joc.0c02820
![]()
![A Path to Substituted Bicyclo[2.1.1]hexanones](https://www.chemistryviews.org/wp-content/uploads/2024/10/1substitutedbicyclo211hexan2ones_2024-125x94.png)
