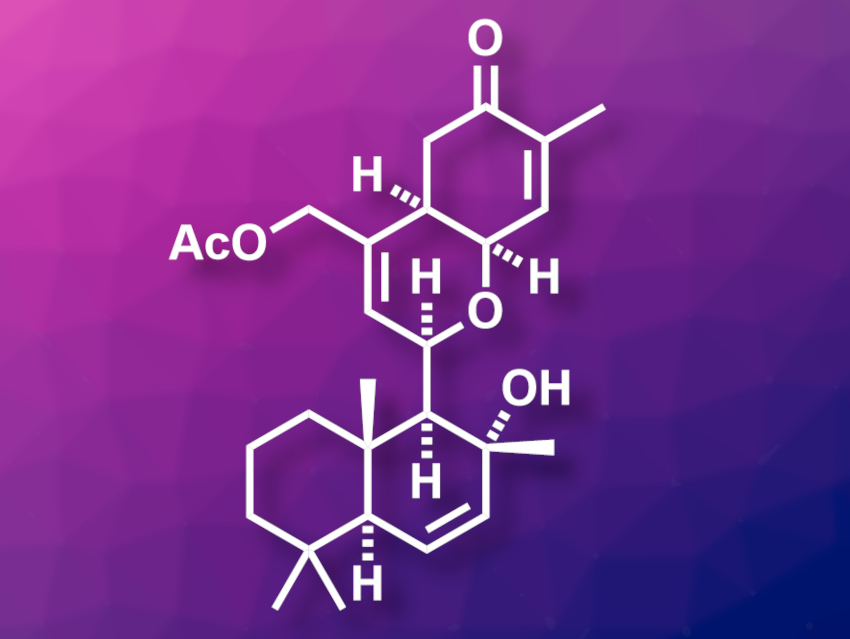
Human immunodeficiency virus (HIV) infections can usually be managed well using antiretroviral drugs. However, this is no cure for HIV because the virus persists in the body and can re-emerge if the treatment is stopped. One strategy towards developing a cure for HIV uses so-called latency-reversing agents (LRAs). LRAs reactivate virus production in latently infected cells and trigger an immune response and/or cell death. Ansellone A (pictured) is a marine sesterterpenoid that shows LRA activity. It has been synthesized in 24 total steps [1], but there is a lack of biological studies of ansellone A and its analogues.
Kenichi Murai, Mitsuhiro Arisawa, Osaka University, Japan, and colleagues have developed a concise synthesis of Ansellone A with 17 total steps and conducted a biological screening of this compound, as well as analogues with other substituents in the place of its –OAc group. The team started from (+)-sclareolide, which was first converted to a diol, then to a triflate-stabilized tertiary allylic alcohol, and finally oxidized to introduce an aldehyde group. The resulting intermediate was subjected to a Prins cyclization reaction with cis-γ-hydroxycarvone to create the tetrahydropyran ring of the target compound. Finally, the triflate group was removed and the acetate substituent was introduced to give ansellone A.
The researchers obtained ansellone A in 17 total steps with a longest linear sequence (LLS) of 13 steps from (+)-sclareolide. They also synthesized analogues with –OH or –OMe groups in place of the acetate substituent for bioactivity studies. The team found that the free alcohol has a higher LRA activity than the parent compound.
- Total Synthesis and Biological Evaluation of the Potent HIV Latency-Reversing Agent Ansellone A and its Analogues,
Mizushi Yanagihara, Kenichi Murai, Naoki Kishimoto, Towa Abe, Shogo Misumi, Mitsuhiro Arisawa,
Org. Lett. 2021.
https://doi.org/10.1021/acs.orglett.1c00151
Reference
- Total Syntheses of Sesterterpenoid Ansellones A and B, and Phorbadione,
Wei Zhang, Hongliang Yao, Jingxun Yu, Zhihong Zhang, Rongbiao Tong,
Angew. Chem. Int. Ed. 2017, 56, 4787–4791.
https://doi.org/10.1002/anie.201701879
![]()