Quinolines and pyridines are important classes of organic molecules. Many examples are bioactive and have applications as pharmaceuticals or agrochemicals. They can also act as ligands in catalysis, and they frequently serve as synthetic intermediates during the synthesis of complex natural products. Pyridines are usually synthesized by cycloadditions or multicomponent reactions, while quinolines are commonly synthesized by annulation reactions. Due to harsh reaction conditions in these syntheses, it is often necessary to introduce the desired functional groups after the construction of the ring system.
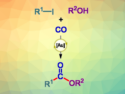
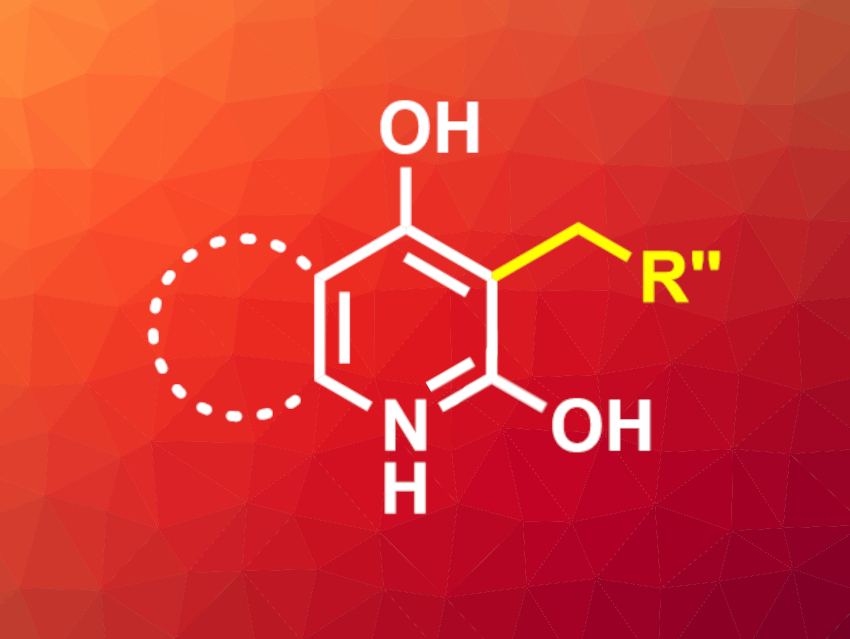
Pratik R. Chheda, Janssen Research and Development, San Diego, CA, USA, and colleagues have developed a reductive C3-alkylation reaction for 2,4-dihydroxyquinolines and -pyridines, mediated by diethyl-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate (Hantzsch ester). The quinoline or pyridine substrate, an aldehyde, and the Hantzsch ester were heated to 100 °C in pyridine for 2–4 h. The desired products (example pictured) were isolated in high yields of up to 92 % by simple precipitation with diethyl ether.
The reaction is compatible with a wide range of aromatic aldehydes containing both electron-donating and electron-withdrawing groups, as well as heteroaromatic aldehydes. Different aliphatic and heteroaliphatic aldehydes were also used successfully. Various dihydroxyquinolines and -pyridines were successfully converted, but no formation of the desired alkylation product was observed when monohydroxyquinolines were used.
The alkylated products were further functionalized by replacing the hydroxy groups with chlorine using POCl3. The chlorinated derivatives can undergo substitution reactions, e.g., with sodium methoxide, azetidine, and dimethylamine. These types of conversions could allow the synthesis of a wide range of substituted quinolines and pyridines.
- One-Pot Reductive Alkylation of 2,4-Dihydroxy Quinolines and Pyridines,
Pratik R. Chheda, David A. Kummer, Rachel T. Nishimura, Kelly J. McClure, Hariharan Venkatesan,
J. Org. Chem. 2021.
https://doi.org/10.1021/acs.joc.1c00496
![]()