The determination of the native structure of bio-molecular systems is often the basis for an understanding of the biological function and mechanism of action. Excepting diffraction methods, no experimental technique is capable per se of providing a quantitative measure of molecular structure. Structural parameters can only be calculated by theoretical methods, which in turn have to be validated by comparing predicted physical properties with experiment.
Ursula Rothlisberger and co-workers, ETH-Lausanne, Switzerland, compare the performance of different computational methods with respect to their ability of reproducing the relative energetics of a medium-sized biomolecule. High level wave function based methods are not applicable, due to their enormous computational costs. Therefore, the team has benchmarked the performance of a diverse, less computationally demanding, set of computational approaches – classical force fields such as AMBER FF96, AMOEBAbio09 and the semiempirical density functional tight (DFT)-binding method.
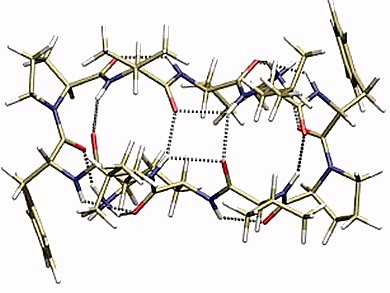
They predict the ground state structure of protonated gramicidin S, a cyclic decapeptide and natural antibiotic against bacteria and fungi, which native structure is unknown.
The DFT method was unique in predicting the correct lowest energy structure, as verified by comparison to experiment. AMOEBA performs the best among the force fields. But none of the major force fields has sufficient accuracy to be used reliably for the energetic prescreening of the candidate structure. Higher-level theory needs to be employed, despite the heavy costs.
- Assessing the performance of computational methods for the prediction of the ground state structure of a cyclic decapeptide,
Manuel Doemer, Matteo Guglielmi, Prashanth Athri, Natalia S. Nagornova, Thomas R. Rizzo, Oleg V. Boyarkin, Ivano Tavernelli, Ursula Rothlisberger,
Int. J. Quantum Chem. 2012.
DOI: 10.1002/qua.24085