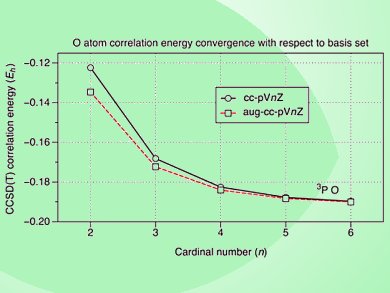
Quantum mechanical calculations involving molecules can be intuitively divided into two parts, the method and the basis set. The choice of the basis set is of fundamental importance because it determines the accuracy of the calculation within the limits of the associated theoretical method. Often it is treated as a black box and selected by experience rather than the understanding of the underlying physics of basis sets. This has severely limited the accessibility of ab-initio calculations for non-specialists and researchers new to the discipline.
J. Grant Hill, University of Glasgow, UK, delves into this question in great detail for Gaussian type orbitals (GTOs). In addition to cover the all-necessary physical and mathematical basis to help new practitioners in their first informed choices of the basis, Grant Hill also discusses issues of computational costs associated with basis sets that account for electron-electron repulsion and electron correlation. These are particularly important matters to be considered in problems involving transition metals, biomolecules, and magnetic materials. In particular, the choice of the correct basis sets for transition metal, heavier alkali and alkaline earth and rare earth elements remains a challenge as these pose problems in terms of strong relativistic effects and multi-reference character.
- Gaussian basis sets for molecular applications,

J. Grant Hill,
Int. J. Quantum Chem. 2012.
DOI: 10.1002/qua.24355
Also of interest:
- Atomic orbital basis sets,
Frank Jensen,
WIREs Comput Mol Sci. 2012.
DOI: 10.1002/wcms.1123