The Time-Dependent density functional theory (TD-DFT) has proven to be the most widely used approach for the study of electronically excited states and optical properties of both organic and inorganic systems. This has led to advances in understanding, e.g., the photochemistry of molecules, the determination of excited state structures, and the emission wavelengths. Further enhancements permit the application to equilibrium (slow) and nonequilibrium (fast) processes, two examples of photophysically critical processes of absorption and fluorescence.
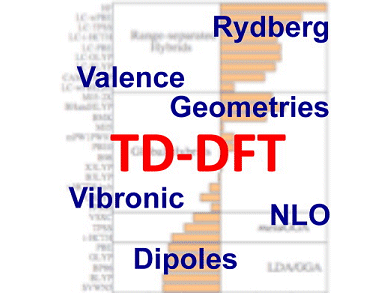
While it is formally an exact theory just like its parent DFT, implementations of the theory require the selection of an exchange correlation functional (XCF) that encapsulates all the necessary approximations. As this choice yields a great impact on the accuracy of the calculated results, one extremely important question for practitioners of TD-DFT is, what is the right TD-DFT functional to use? The answer obviously depends on the problem (specific system and the property of interest) being studied.
To help researchers navigate this fundamental choice, Adèle Laurent and Denis Jacquemin, Université de Nantes, France, have compiled a comprehensive list of high-performance functionals while providing some interesting rules of the thumb for their use. For example, they find that for the majority of properties, states and molecules, pure density functionals that do not incorporate exact exchange (EXX), provide poorer estimates than hybrid functionals. In some cases, such as the energy and geometry of charge-transfer excited states, pure XCF yield dramatic qualitative failures. The only compounds for which EXX-free XCF can be really satisfying are very compact molecules.
This fundamental benchmark is expected to help researchers in their task of predicting atomic and molecular spectra and charge transfer processes, which are critical to an understanding of photochemistry of molecular systems and the rational design of corresponding solar cells.
- TD-DFT benchmarks: A review,
Adèle D. Laurent, Denis Jacquemin,
Int. J. Quantum Chem. 2013.
DOI: 10.1002/qua.24438