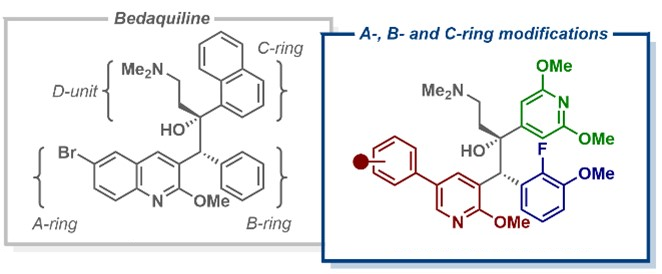
Tuberculosis (TB) was first classified as a global health emergency almost 30 years ago and remains an important cause of death. In response to the emergence of drug-resistant TB variants, in 2012, a new anti-TB drug called bedaquiline was granted accelerated approval from the U.S. Food and Drug Administration (FDA). Despite being highly effective against drug-resistant TB, the clinical use of bedaquiline (pictured below) has been limited due to safety concerns. For example, it could lead to cardiac issues due to inhibition of the hERG cardiac potassium ion channel.
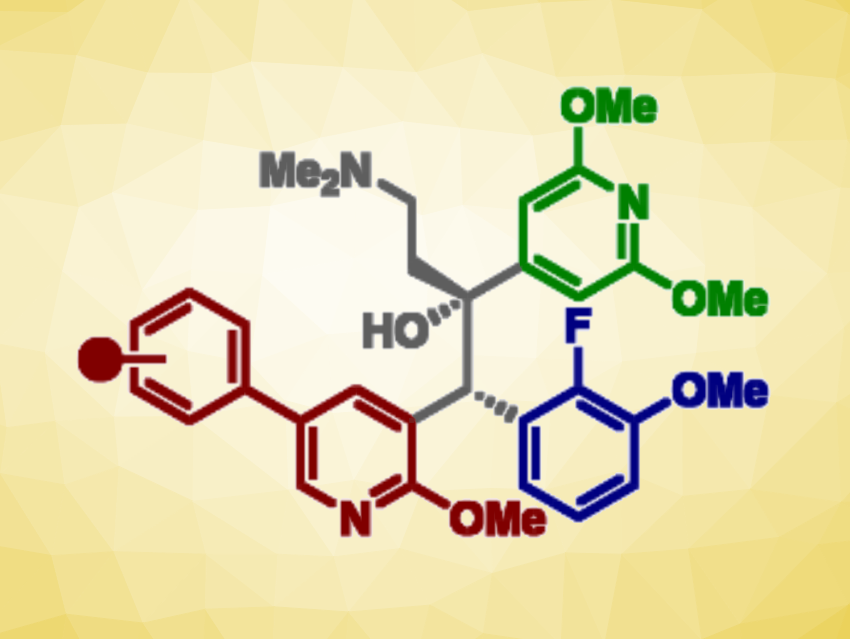
Building on the observation that anti-TB activity was retained when bedaquiline’s quinoline ring was replaced with a phenylpyridine scaffold, Daniel L. Priebbenow, Monash University and The University of Melbourne, Parkville, Australia, Jonathan B. Baell, Monash University, and colleagues have designed and synthesized a new series of bedaquiline analogues. The team modified each aryl region of bedaquiline (pictured below, A-, B-, and C-ring) and combined the phenylpyridine scaffold at the A-ring site with optimized structural elements at the B- and C- ring, as in the promising anti-TB candidate TBAJ-587.
The team found that 4-trifluoromethoxyphenyl and 4-chlorophenyl pyridyl analogues of TBAJ-587 retained relatively potent anti-tubercular activity. With the 4-chlorophenyl derivative, in particular, a significant reduction in hERG inhibition compared with bedaquiline was achieved. Ultimately, these modifications led to the discovery of less lipophilic, potent anti-TB compounds with a reduced risk of toxicity. These findings could be helpful for the future development of safer anti-TB therapeutics inspired by bedaquiline.
- Discovery of Anti‐tubercular Analogues of Bedaquiline with Modified A‐, B‐ and C‐Ring Subunits,
Lisa Barbaro, Gayathri Nagalingam, James A. Triccas, Lendl Tan, Nicholas P. West, Daniel Priebbenow, Jonathan B. Baell,
ChemMedChem 2022.
https://doi.org/10.1002/cmdc.202200533
![]()