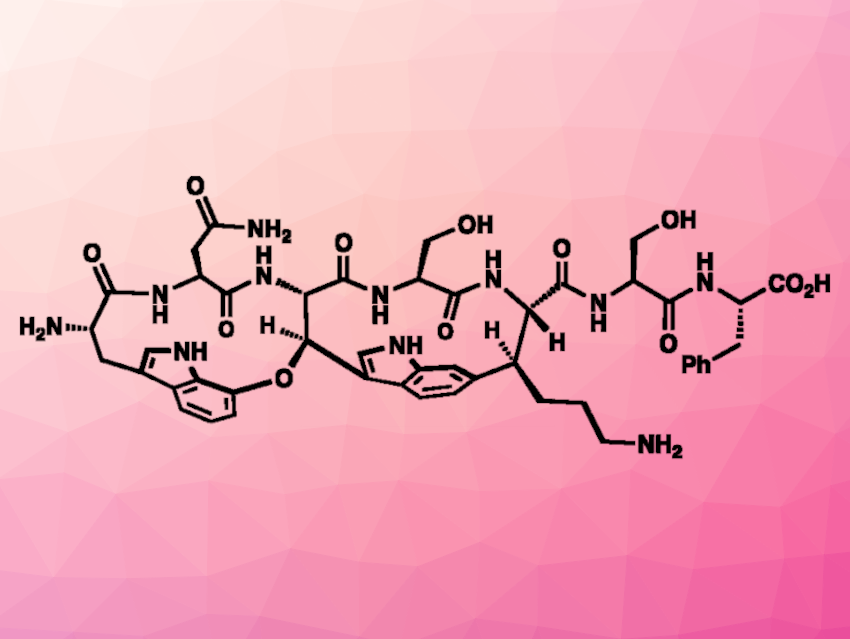
New types of antibiotics are urgently needed due to the emergence of multidrug-resistant pathogens. Darobactin A (pictured), for example, has shown antibiotic activity against Gram-negative pathogens. It was isolated from Photorhabdus bacteria, and its structure features a highly strained fused ring system based on macrocyclic peptides. It has a rigid structure that is considered to be important for its activity. To study the antibiotic properties of darobactin A and potentially use it for drug development, a viable total synthesis of the compound would be useful.
Phil S. Baran, Scripps Research, La Jolla, CA, USA, and colleagues have performed a modular total synthesis of darobactin A. The team used commercially available amino acids and arenes as building blocks for the synthesis. They first prepared two unnatural amino acids using decarboxylative alkynylations as key steps. The resulting unnatural amino acids were connected to arene-based intermediates and subjected to another key reaction: a Larock cyclization to close the five-membered rings of the target compound’s central indole unit. The desired product was obtained in an atroposelective manner. The same type of reaction was used to build the second indole and close the second ring of darobactin A.
Further transformations at the side chains then gave the desired product. The synthetic strategy was designed with further medicinal chemistry studies in mind and allows the preparation of analogues of darobactin A that could be useful for drug development.
- Atroposelective Total Synthesis of Darobactin A,
You-Chen Lin, Fabian Schneider, Kelly J. Eberle, Debora Chiodi, Hugh Nakamura, Solomon H. Reisberg, Jason Chen, Masato Saito, Phil S. Baran,
J. Am. Chem. Soc. 2022.
https://doi.org/10.1021/jacs.2c05892
![]()