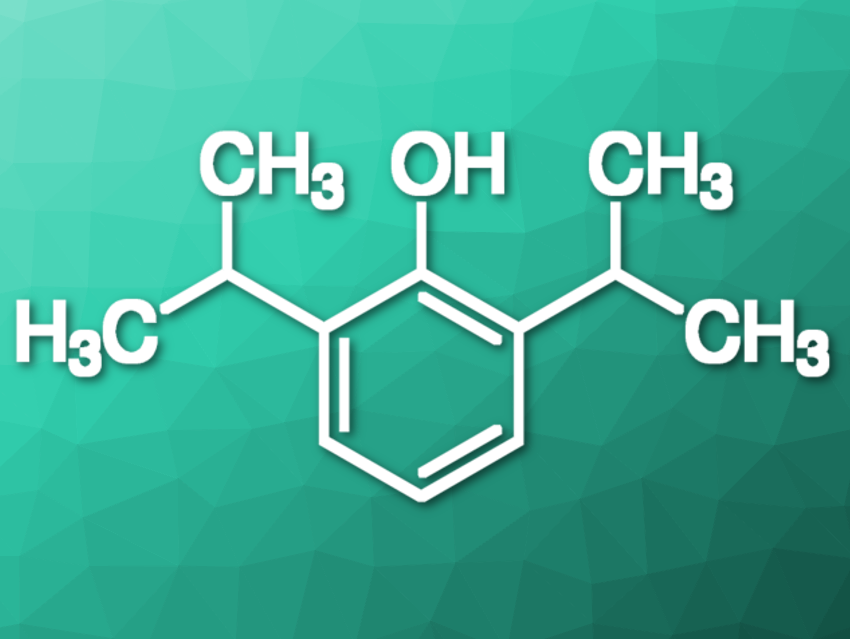
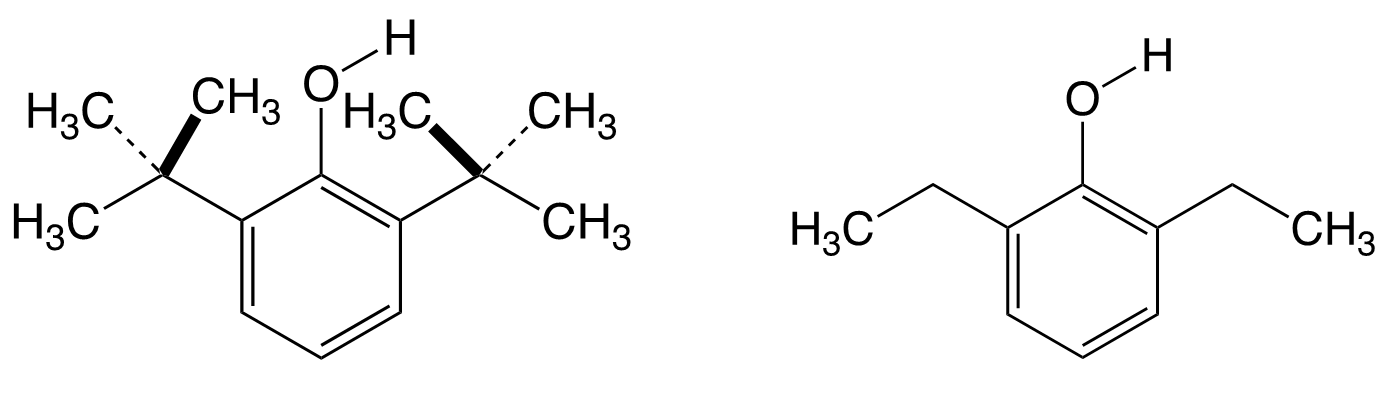
The molecule I have been fascinated with for over a quarter of a century is quite simple: 2,6-diisopropylphenol. Earlier studies on the reactivity of phenolic O–H revealed a steric strain in 2,6-di-tert-butylphenol (pictured below on the left). No such strain was found for 2,6-diethyl substituents (pictured below on the right). I was particularly fascinated by the structure of 2,6-diisopropylphenol because the question arose: What kind of influence would the 2,6-diisopropyl substituents exert on the O–H group? Since the free or hindered character of the O–H bond is reflected by the infrared spectra, I turned to spectroscopy.
1975 Infrared Spectrum of 2,6-Diisopropylphenol
The infrared (IR) spectrum of 2,6-diisopropylphenol in dilute CCl4 solution shows absorptions for the O–H stretching region at 3619 (intense peak) and 3641 cm–1 (shoulder). The absorption peak is shifted towards frequencies higher than 3640 cm–1 for 2,6-tert-butyl-phenols, as evidence of a steric strain on the O–H bond.
Comparison to 2-tert-Butyl-Substituted Phenols
The IR spectrum of 2-tert-butyl-substituted phenols shows two absorption peaks. This leads to the reasonable assumption that the O–H groups of the phenols are coplanar with the aromatic ring due to hindered rotation. Therefore, in 2-tert-butyl-phenol, the O–H is either cis or trans to the substituent (pictured below), resulting in two different absorptions at 3647 cm–1 and 3608 cm–1, respectively. The presence of cis and trans isomers could also be observed when the 6-position of the 2-tert-butylphenol was substituted by a methyl group [1].
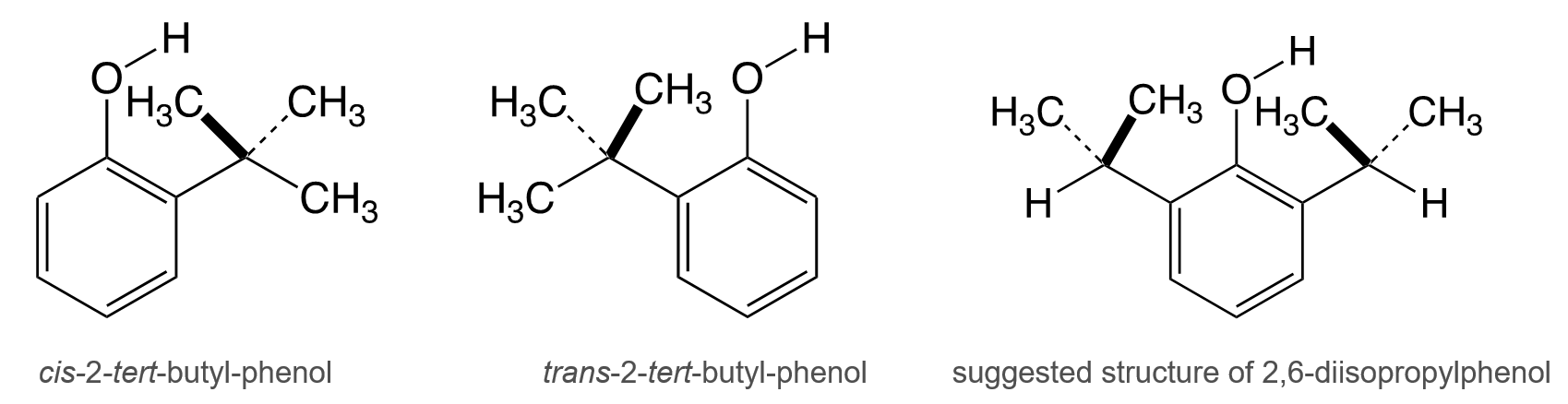
These findings tentatively led to the assumption that the origin of the 3641 cm–1 peak of 2,6-diisopropylphenol was caused by the steric interaction of the O–H with the specific conformer of the isopropyl group in which the O–H is “captured” between two methyl groups of the isopropyl substituent (pictured above) [1].
Quantum Chemical Calculations Lead to the Solution in 2001
A more detailed study of the problem indicated that the absorption peaks at 3619 cm–1 and 3641 cm–1 could not be satisfactorily reproduced by two symmetric band components. Instead, ab initio quantum chemical calculations were used to identify five stable conformers that differ in the relative orientations of the two ortho-substituents.
The experimental spectrum could be successfully simulated by these calculated frequencies and band intensities. The band intensities could be assigned to the five conformers and their relative stability [2].
References
[1] M. Simonyi, I. Kovács, J. Kardos, S. Holly, “Steric” absorption peak in the infrared spectra of 2,6-diisopropylphenols, Tetrahedron Lett. 1975, 16, 1631–1632. https://doi.org/10.1016/S0040-4039(00)72217-9
[2] Z. Bikádi, G. Keresztury, S. Holly, O. Egyed, I. Mayer, M. Simonyi, Role of Secondary Interactions in the Conformational Equilibrium of 2,6-Diisopropylphenol, J. Phys. Chem. A 2001, 105, 3471–3474. https://doi.org/10.1021/jp0030387
Author
Dr. Miklos Simonyi, Research Center for Natural Sciences, Budapest, Hungary
- Your Molecule Story
Share a molecule and why it is special to you and have the chance to win a book
![]()



