
After covering saccharin, the poisonous lead(II) acetate, and cyclamate, we look at another low-calorie sweetening agent: aspartame.
14. Aspartame
Discovery
James Schlatter, a chemist with the firm G. D. Searle & Company, Skokie, IL, USA, was charged with finding new possibilities for treating stomach ulcers. In the context of this project, in December 1965, he decided to prepare the tetrapeptide H-Trp-Met-Asp-Phe-OH. This particular amino-acid sequence corresponds to the C-terminus of the enzyme gastrin, which among other things intensely stimulates the production of stomach acid.
In the course of the multistep synthesis, one of the intermediates generated was the methylated dipeptide H-Asp-Phe-OCH3 (5), a compound that had recently been synthesized for the first time by J. S. Morley’s research group in England [52]. But Schlatter had the good luck to have a mishap in the course of recrystallizing the compound [53], which in turn helped him make a sensational discovery:
“As I heated the compound in a flask with methanol, some of the contents splashed on the outside of the flask. That’s how a bit of material came to be on my finger. Somewhat later, as I happened to lick my fingers in order to pick up a thin sheet of weighing paper, I noticed an intense sweet taste. My first thought was that I still had some sugar on my fingers from breakfast. At any rate, it soon became clear to me that this couldn’t be the case after all, since in the meantime I had washed my hands. So I followed the trail of the substance on my hand backward to the flask in which I had recrystallized the aspartyl phenylalanine methyl ester. It was clear to me that a dipeptide ester couldn’t be toxic, so I tasted a little of it deliberately, and found that it really was the same sweet substance I had previously detected by accident on my finger.”
|
|
|
Figure 17. Synthesis of aspartame. |
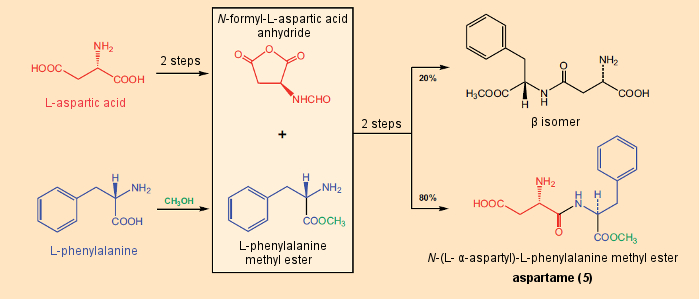
That this substance would be sweet was not predictable, since neither L-aspartic acid nor L-phenylalanine has a sweet taste; the latter, in fact, is bitter. From this N-L-α-aspartyl-L-phenylalanine-1-methyl ester (5), later to become familiar under the name aspartame, the three stereoisomers with the alternative Asp-Phe configurations L/D, D/D, and D/L were synthesized, and all three were found to have bitter tastes [54, 55]. Further systematic structural variations showed that every sweet-tasting peptide in the series had on its N terminus an L-aspartic acid with free α-amino acid and γ-carboxyl groups [56].
Within a short period of time, hundreds of relevant peptides had been prepared, from among which only a very few had higher sweetening power than aspartame. From a commercial standpoint, however, none of the latter came into question, since their syntheses (see Fig. 17) were relatively more complicated, causing them to be more expensive, or else they contained unusual amino acids, so that their physiological degradation was less well known relative to substances like the endogenous amino acids phenylalanine and aspartic acid.
Today, aspartame is prepared by direct reaction of the methyl ester of phenylalanine with N-formylated aspartic acid anhydride. A byproduct formed in 20 % yield, the β-Isomer from “incorrect” peptide coupling, is removed by recrystallization [57].
Tolerance
As in the cases of saccharin and cyclamate, it was true with aspartame as well that initial tolerance tests with rats led at first to controversial discussions regarding its physiological harmlessness. Preliminary approval was granted for a few months in 1974 and 1975, but this was soon withdrawn again. It was not until 1981 that G.D. Searle & Company received a lasting permit for aspartame (NutraSweet®, Equal®), albeit initially only for use in “dry products” (e.g., cereals). In the United States, aspartame was further approved in 1983 for use in carbonated beverages, and in 1990 as a sweetener for food products generally.
After further tolerance studies, toward the end of 1984 even the FDA and the Centers for Disease Control, followed in 1985 by the American Medical Association, agreed unanimously that aspartame did not represent any risk to health. In addition, the European health authorities unanimously adopted the position that up to an ADI-value of 40 mg/kg body weight/day, aspartame is completely harmless from a health standpoint [58].
Metabolism
In contrast to saccharin and cyclamate, aspartame, being a dipeptide, has a nutritional value comparable to that of sugar. However, because of its high sweetness value, the amount of aspartame consumed is so small that this sweetener, too, can be regarded as essentially free of calories. Nevertheless, when compared to saccharin and cyclamate, aspartame has one distinctive drawback: it is less stable. The chemical reason for this is the acid-lability of the ester and peptide bonds, and the typical ring-closure reaction of amino acids to dioxopiperazines (see Fig. 18). Through thermal and acid-catalyzed decomposition, aspartame undergoes methanol elimination. In the course of complete digestion in the human gastrointestinal tract, both the ester linkage and the peptide bond between phenylalanine and aspartic acid are cleaved.
|
|
|
Figure 18. Decomposition of aspartame. |
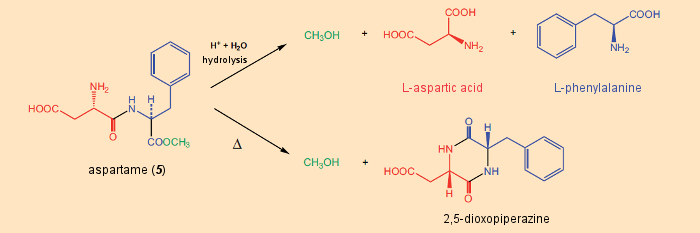
In the solid state, aspartame is subject to slow decomposition at temperatures above 120 °C [59]. In aqueous solution, the rate of decomposition is a function of time, temperature, and pH (see Fig. 19). Fortunately, its optimum stability occurs at pH = 4 [60], and thus in the normal region for colas and other soft drinks. It is here, as well as in its outstanding taste characteristics, that the basis for aspartame’s spectacular success is to be found.
|
|
|
Figure 19. Stability of aspartame. Left: Half-life of aspartame at 25 °C as a function of pH; right: Decomposition of aspartame at 80 °C as a function of pH. |
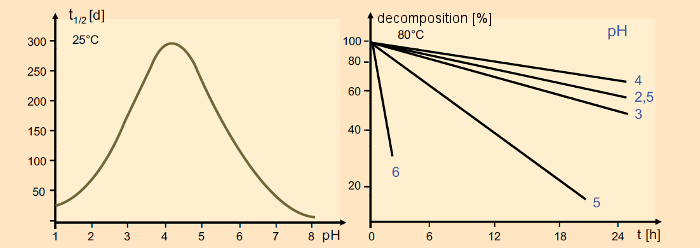
The lability of the ester and peptide bonds in the digestive tract, but also through inappropriate storage or processing (e.g., after lengthy boiling), leads to the decomposition products methanol, phenylalanine, and aspartic acid. The high toxicity of methanol is well known and is a consequence of its oxidation to formaldehyde and formic acid.
Nevertheless, there is no risk of methanol poisoning from aspartame, since the amount released is extremely small, and even consumption of large amounts of aspartame causes almost no change in the concentration of methanol in the blood. By comparison, drinking similar amounts of orange juice, apple juice, and especially tomato juice can release as much as the five-fold quantity of methanol into the human body [61, 62].
An all-clear signal can also be given for digestion of aspartame with respect to the natural amino acids phenylalanine and aspartic acids, both of which are released upon decomposition. This generalization does not apply, however, to those persons suffering from phenylketonuria (PKU). In the case of this relatively common genetic disorder (1 in 8000 newborns), phenylalanine cannot be transformed into the important amino acid tyrosine due to the absence of the corresponding enzyme, phenylalanine hydroxylase. The condition is revealed through urine tests by the presence of the unusual degradation products, phenylketones, which are the source of the ailment’s name.
Untreated, PKU leads to serious mental developmental disorders as well as epilepsy. In many countries, newborns are routinely scanned for this problem; serious mental handicaps can be prevented by strict observance of a diet with rigorous limitations on egg white intake during the growth phase. Due to the potential risk to these patients, all foods containing aspartame must be provided with a warning label.
Popularity
Despite the long delay between the discovery of aspartame in 1965 and its certification in 1981 as a permissible sweetener in the United States (from 1983 also acceptable in carbonated beverages), aspartame profited significantly from the fact that it could be seen chemically as a fragment of a natural protein. From the very outset, this sweetener was viewed with significant confidence, more at least than for the banned cyclamate in the United States, as well as for saccharin, which has continued to be associated with suspicions of carcinogenicity. When aspartame was first permitted for use in carbonated beverages, both Diet Pepsi and the newly introduced Diet Coke [63] were sweetened exclusively with saccharin, still wrongfully suffering from a bad reputation. But in 1983, both began instead to introduce aspartame into their respective calorie-free products.
At first, Diet Coke switched to a saccharin/aspartame mixture, but Diet Pepsi was reformulated immediately to use of aspartame exclusively. Pepsi, of course, exploited this step to the fullest in their advertising strategy (see Fig. 20, left), but ultimately in vain, because Coca-Cola soon adapted their own recipe to conform (see Fig. 20, right). In the long run, that paid off: in 2011, Diet Coke took over second place on the hit-list of American soft drinks, surpassed only by the sugar-containing “original” Coca-Cola.
|
|
|
Figure 20. Aspartame in softdrinks (photo left: OMNI Magazine, 1985; photo right: Dave Allen). |

References
[52] H. Gregory et al., J. Chem. Soc. (C) 1966, 522–531. DOI: 10.1039/J39680000522
[53] Lewis D. Stegink, Lloyd J. Filer (Eds.), Aspartame: Physiology and Biochemistry, Marcel Dekker, New York, USA, 1984. ISBN: 978-0-8247-7206-2
[54] R. H. Mazur et al., J. Am. Chem. Soc. 1969, 91, 2684–2691. DOI: 10.1021/ja01038a046
[55] R. H. Mazur et al., J. Med. Chem. 1973, 16, 1284–1287. DOI: 10.1021/jm00269a014
[56] D. E. Walters, F. T. Orthoefer, G.E. DuBois (Eds.), Sweeteners, ACS Symposium Series, American Chemical Society, Washington, D.C., USA, 1991. DOI: 10.1021/bk-1991-0450
[57] G.-W. von Rymon Lipinski, in Ullmann’s Encylopedia of Industrial Chemistry, 7th edition, Wiley-VCH, Weinheim, Germany, 2011. DOI: 10.1002/14356007
[58] European Commission, Update on the Safety of Aspartame, 2002. Link
[59] S. Rastogi et al., Pharm. Res. 2001, 18, 267–273. DOI: 10.1023/A:1011086409967
[60] A. Ripper et al., in Alternative Sweeteners, Marcel Dekker, New York, USA, 1986. ISBN: 978-0-8247-7491-2
[61] K. Wucherpfennig et al., Flüssiges Obst, 1983, 8, 348–354 (in German).
[62] E. G. Abegaz et al., in Alternative Sweeteners, 4th edition, CRC Press, Boca Raton, USA, 2012. ISBN: 978-1-4398-4615-5
[63] M. Pendergast, Für Gott, Vaterland und Coca-Cola, Zsolnay, Vienna, 1993 (in German). ISBN: 978-3-552-04624-5
The article has been published in German as:
- Die Saccharin-Saga,
Klaus Roth, Erich Lück,
Chem. unserer Zeit 2011, 45, 406–423.
DOI: 10.1002/ciuz.201100574
and
- Kalorienfreie Süße aus Labor und Natur,
Klaus Roth, Erich Lück,
Chem. unserer Zeit 2012, 46, 168–192.
DOI: 10.1002/ciuz.201200587
and was translated by W. E. Russey.
The Saccharin Saga – Part 1
The invention of the first artificial sweetener and a lifetime battle for credit
The Saccharin Saga – Part 2
The early industrial production and organized smuggling of saccharin
The Saccharin Saga – Part 3
The health concerns associated with artificial sweeteners
The Saccharin Saga – Part 4
A glance back to ancient Rome, and the most hair-raising of all sweeteners
The Saccharin Saga – Part 5
What’s in your softdrink? – Introducing cyclamate
The Saccharin Saga – Part 6
Aspartame – a sweet dipeptide ester
The Saccharin Saga – Part 7
Acesulfame-K – another successful sweetening agent
The Saccharin Saga – Part 8
Thaumatin – a sweet protein with a licorice aftertaste
The Saccharin Saga – Part 9
Sucrose or Splenda turned into a low-calorie alternative to sucralose or saccharose
The Saccharin Saga – Part 10
Combining sweet cations and anions, and turning bitter compounds into sweeteners
The Saccharin Saga – Part 11
Intelligent synthetic strategies for low-calorie sweeteners
The Saccharin Saga – Part 12
Stevia plant extracts as low-calorie sweeteners
The Saccharin Saga – Part 13
Finding the best mixture of sweeteners to replicate the taste of real sugar
See all articles by Klaus Roth published in ChemViews Magazine



